
通過顯性突變,氨酰化tRNA合成酶構成了與CMT疾病相關的最大的蛋白家族。其中一個例子是CMT亞型2N(CMT2N),由分布在丙氨酰tRNA合成酶(AlaRS)中的單個突變導致,其中包括三個位于氨酰化結構域中的突變,因此表明tRNA裝載缺陷產生的一種作用。然而,我們發(fā)現其中兩個突變導致氨酰化缺陷,但是在體外純化的蛋白和在病人的細胞樣本中,最廣泛存在的R329H突變在氨酰化功能上都是正常的。值得注意的是,與野生型丙氨酰tRNA合成酶相比,所有三個突變蛋白均具備了與Nrp1蛋白相互作用的能力,Nrp1先前被證明與甘氨酰tRNA合成酶導致的CMT病變相關。異常的AlaRS與Nrp1的相互作用在攜帶有R329H突變的病人樣本中得到進一步的確認。然而,在氨酰化域之外的CMT2N突變并不會誘導Nrp1的相互作用。詳盡的生物化學和生物物理學檢測技術,包括X-射線晶體衍射,小角X射線散射,氫氘交換(HDX),switchSENSE流體動力學檢測技術和蛋白酶消化表明突變誘導氨酰化域在結構上的松弛性,這種松弛的結構與Nrp1的相互作用相關。Nrp1的b1b2域與R329H AlaRS的互作相關。結果表明,Nrp1廣泛的與CMT相關的tRNA合成酶家族成員發(fā)生互作。此外,我們發(fā)現一個明顯的由在校對域產生的突變導致的結構松弛,但是在C-Ala域的突變對結構沒有影響,表明相同蛋白的突變可能通過不同的機制導致神經病變。我們的結果表明,如同其他CMT相關的tRNA合成酶,氨酰化本身與病變并不相關。
CMT是最常見的遺傳性神經病變,全球患病率為1/2500,除了支持性護理,目前沒有可行性治療方法,也被稱為遺傳性運動和感覺神經性病變,該疾病主要影響控制肌肉運動以及傳遞信息至大腦的外周神經,導致了肌無力以及感覺喪失,尤其是作用于手和腳。從遺傳上來看,CMT是一組與編碼100個蛋白的基因相關連的疾病。雖然對于某些基因它們與外周神經結構和/或功能的關聯(lián)是明顯的,但對于其它許多基因來說,情況并非如此。在這些基因中,最主要的是編碼氨基酸酰基轉移核糖核蛋白(tRNA)合成酶(aaRSs)。到目前為止,五個aaRSs(例如glycyl [GlyRS or GARS1]-, tyrosyl [TyrRS or YARS1]-, histidyl [HisRS or HARS1]-, alanyl [AlaRS or AARS1]-和 tryptophanyl [TrpRS 或 WARS1]-tRNA synthetases)經證實與疾病相關,構成了導致CMT發(fā)生的最大基因家族。
aaRSs是進化上保守的,負責tRNA與其同源氨基酸相結合的必須的酶,以支持核糖體上蛋白的合成。在復雜的多細胞生物體內,aaRSs的功能已經超出了本身的酶活性功能而具有了廣泛的調節(jié)功能。至少對于三個CMT-相關的aaRSs,包括 GlyRS, TyrRS和HisRS,功能喪失性機制已經被排除了。部分是基于CMT相關的aaRSs中的突變是顯性的,病人總是具有一個受影響和一個未受影響的等位基因,表明即使突變存在缺陷,突變的aaRS與野生型的酶共同支持tRNA的氨酰化。有趣的是,在這三個aaRSs中,導致CMT發(fā)生的突變都誘導了蛋白結構的松弛,這種結構的松弛使得翻譯機制之外的異常互作發(fā)生,這種互作對疾病的發(fā)生起著至關重要的作用。尤其,由分泌的CMT突變型GlyRS與跨膜受體神經纖毛蛋白1(Nrp1)的相互作用抑制了VEGFA與該受體的結合和傳遞神經信號。
AlaRS是aaRSs家族中與CMT強關聯(lián)的蛋白之一,其導致的CMT被稱為亞型2N(CMT2N),也被稱為AD-CMTax-AlaRS,以表明其為常染色體顯性遺傳且以軸索型癥狀為主。與其他CMT-相關的以二聚體形式發(fā)揮作用的aaRSs相反,人的AlaRS以單體形式催化tRNA氨酰化反應。AlaRS具有的這種特定的特征非常有意思,因為已經表明對于那些二聚體形態(tài)的aaRSs(例如GlyRS, TyrRS和HisRS),CMT突變誘導的結構松弛主要發(fā)生在二聚化的互作界面。那么,AlaRS中導致CMT發(fā)生的突變也會導致結構的松弛嗎?如果是,結構松弛發(fā)生位點是在哪里呢?
AlaRS在CMT相關的aaRSs中也是獨一無二的,因為它包含一個校準域,位于N端的氨酰化域和C端的C-Ala域之間(請見Fig. 1A)。AlaRS的氨酰化域對于tRNA和其同源氨基酸丙氨酸的結合是必要且充分的。它不僅包含催化活性位點同時也包括關鍵的負責識別tRNAAla的主要特性元件,這是一個位于接受鏈莖段中的 G3:U70 型錯配堿基對。然而由于丙氨酸和其他氨基酸,例如絲氨酸和谷氨酸,的高度相似性,AlaRS的活性位點在選擇同源氨基酸方面是欠缺的。因此,一個校準域被引入AlaRS中,以糾正這種發(fā)生的錯誤。AlaRS水解校準功能的重要性已經得到廣泛的證實,因為哪怕是最輕微的校準缺陷將會導致嚴重的疾病。人的AlaRS的C-Ala域的功能還不清楚。在原核AlaRS中,C-Ala幫助tRNA的結合,因此提高了酶的活性,然而,人的AlaRS失去了該功能。
促進CMT發(fā)生的突變在三個結構域中已經得到證實。為了廣泛的研究CMT突變對AlaRS結構和功能的影響,我們納入了每個結構域的突變,包括在氨酰化結構域的三種突變(N71Y, G102R和R329H),在校準域的一種突變(E688G),和在C-Ala結構域的兩種突變(E778A和D893N)(請見Fig. 1A和B)。在它們之間,位于氨酰化域的R329H突變當前已在來自于四個不同國家的八個不相關的家族中被鑒定,表明這種突變超強的促疾病發(fā)生的能力。我們發(fā)現僅僅位于氨酰化域的N71Y和G102R突變,而并非R329H突變或校準域的突變和C-Ala域的突變,損害了催化活性。
一致的是,在攜帶R329H突變的病人樣本中,沒有檢測到tRNA氨酰化缺陷,確定了酶活性功能的喪失并不是CMT2N發(fā)生的原因。沒有一個突變,包括位于校準域的突變E688G影響AlaRS的校準活性,表明CMT2N與校準域的功能缺陷也沒有關聯(lián)。氨酰化和校準域的突變在該結構域誘導了一個局部性的結構松弛變化,然而在C-Ala域的突變沒有構象上的影響。有趣的是,不論是否對酶的活性產生影響,在氨酰化域的所有突變都導致了一個異常的,功能獲得性的與Nrp1蛋白的相互作用。這種異常的AlaRS-Nrp1的相互作用在攜帶有R329H突變的病人樣本中進一步被證實。這表明該突變產生的功能獲得性影響與酶活功能是分開的。此外,該研究提供了證明,表明在不同的tRNA合成酶中,一個非酶活的功能獲得性效應也可能是一個共同的疾病發(fā)生機制。
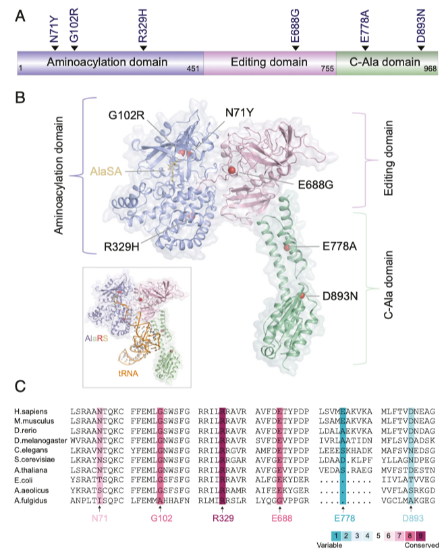
Fig 1 在AlaRS中促進CMT發(fā)生的突變分布
CMT2N突變不影響AlaRS
的單體狀態(tài)或蛋白的穩(wěn)定性
我們首先明確CMT突變是否影響AlaRS的單體狀態(tài)。重組的帶有His-標簽的AlaRS野生型和六個CMT突變蛋白在大腸桿菌中過表達和純化。所有的蛋白都具有相似的產出,表明CMT突變不影響AlaRS的穩(wěn)定性。凝膠過濾層析法分析確認人的AlaRS主要作為單體存在,CMT突變不改變AlaRS的單體狀態(tài),我們也進行了基于熒光的熱遷移分析法(TSAs)。所有蛋白的溶解溫度(Tm)在1度之內,除了N71Y,它在溶解溫度上增加了2.3度,表明更高的穩(wěn)定性(請間Fig. 2A)。因此CMT突變不影響蛋白的穩(wěn)定性。
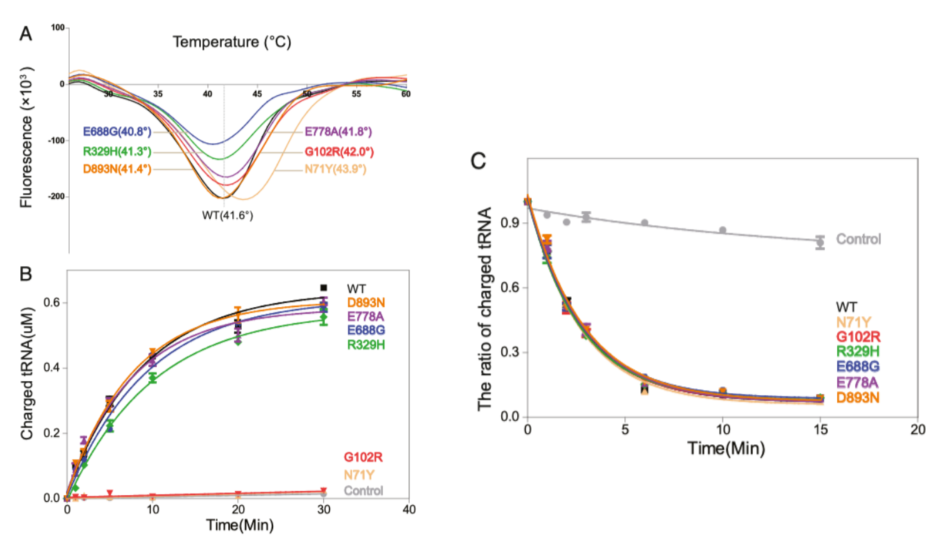
Fig 2 對 AlaRS 蛋白及其 CMT 突變體的酶學分析
包括R329H在內的大多數
CMT2N突變不影響tRNA的氨酰化
AlaRS催化的氨酰化反應需要兩步。第一步,丙氨酸在AlaRS的激活位點被激活,形成一個與酶相連的Ala-AMP中間產物;在第二步,激活的氨基酸被轉移到tRNA的3‘端,產生氨酰-tRNA產物,釋放AMP。為了研究CMT突變對tRNA裝載氨基酸的影響,我們進行了體外的氨酰化檢測,其通過純化重組的AlaRS蛋白檢測了總的反應效率以及體外轉錄的tRNAAla。雖然N71Y和G102R,兩個都位于AlaRS的催化位點附近(請見Fig. 1B),完全消除了酶活性,來自于氨酰化域(但遠離酶活性位點)的R329H突變和來自于校準域的突變(E688G)以及C-Ala域的突變(E778A和D893N)對酶活性都具有較小的影響(請見Fig. 2B)。
攜帶有R329H突變的CMT
病人在tRNA氨酰化功能上沒有缺失
由于R329H突變當前是在CMT病人中被鑒定出,并且先前研究表明屬于功能喪失性突變,我們檢測了攜帶這種突變的CMT2N病人的淋巴細胞。蛋白免疫印跡分析表明與健康個體相比,在三個不同的CMT突變病人中,AlaRS的蛋白水平都沒有明顯的改變,明確了蛋白的穩(wěn)定性(請見Fig. 3A)。通過體外的氨酰化檢測,在CMT病人細胞的裂解液中,我們未檢測到酶活性的缺失(請見Fig. 3B)。最后,Northern blot分析明確了病人中內源性tRNA Ala氨酰化效率的缺失(請見Fig. 3C和D)。
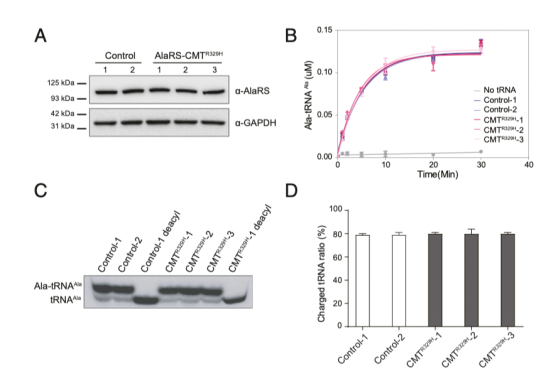
Fig. 3 攜帶有AlaRS R329H突變的CMT病人細胞在tRNA氨酰化功能上沒有缺失
氫-氘交換分析表明CMT突變
對AlaRS構象的不同影響
AlaRS-R329H對蛋白的水解呈現明顯的易感性表明CMT突變增加了蛋白結構的靈活可變性,使人想起在其他CMT相關的aaRSs中所觀察到的現象。為了明確指出全長蛋白狀態(tài)下構象改變的位點,并比較不同的CMT突變,我們對所有六個CMT突變進行了氫-氘交換質譜(HDX-MS)分析,并且將野生型的AlaRS作為參考。當一種蛋白的溶劑從H2 O換成D2 O時,蛋白骨架上的酰胺氫原子經歷了氫-氘交換過程。交換率,或者氘吸收,代表了溶劑的可進入性以及蛋白上每個區(qū)域的構象動力學。在結構上,突變誘導的構象改變和動力學能夠通過比較突變和野生型蛋白進行推測。除了在C-Ala域的兩個突變,與野生型AlaRS相比,位于氨酰化域的突變(N71,G102R和R329H)和校準域的突變(E688G)都表明在氘吸收上的整體增加(請見Fig. 4A)。構象改變的位點,如果有的話,是被限制在攜帶突變的區(qū)域。特別的是,位于氨酰化域的突變僅僅增加氨酰化域的靈活性,校準域的突變僅僅增加校準域的靈活性,然而沒有檢測到由C-Ala域突變導致的構象變化(請見Fig. 4B)。在氨酰化域,G102R和R329H在突變位點附近誘導了更強的局部構象改變,然而,N71Y的影響分布更為廣泛(請見Fig. 4B)。與我們在晶體過程中和水解分析中觀察到的一致,R329H突變促進了氨酰化域C端構象靈活性的增加(請見Fig. 4A)。
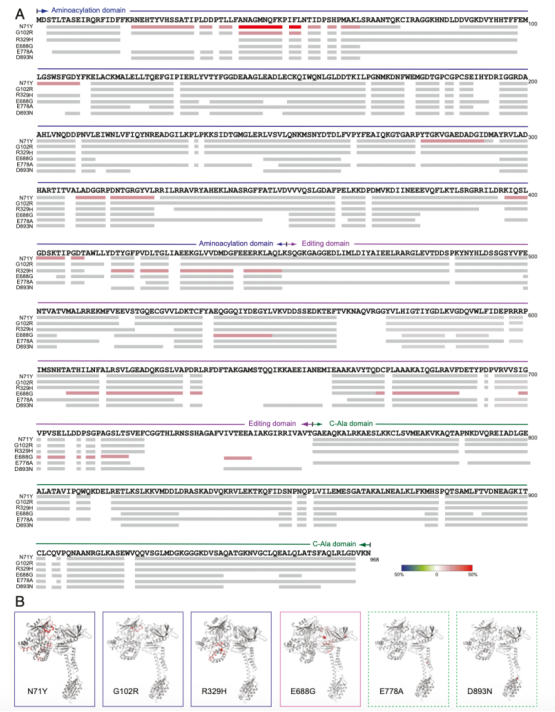
Fig. 4 人類 AlaRS 基因導致先天性肌張力障礙的突變的 HDX 分析
小角度X射線散色明確了在氨酰化域
和校準域的突變導致的結構松弛性
為了明確CMT突變對AlaRS構象的不同效應,我們對AlaRS蛋白進行了小角度散色(SAXS)分析。對于每一個蛋白樣本,散色曲線的形狀獨立于蛋白濃度,并且相應的Guinier曲線展示了平行和線性擬合,表明在測量過程中未發(fā)生明顯的聚集。從散射曲線推測出的分子質量大約都是110kDa,與所有的AlaRS蛋白的單體狀態(tài)一致。從頭開始的形狀重構顯示,與野生型的AlaRS和C-Ala域的突變相比,氨酰化和校準域的突變導致了更加延伸的分子形狀(例如N71Y,G102R,R329H和E688G)(請見Fig. 5A)。與此一致的是,除了 C-Ala 結構域中的兩個突變體外,從散射數據得出的所有突變體的回轉半徑和最大尺寸(Dmax)均大于野生型丙氨酰-tRNA 合成酶(請見Fig. 5B)。有趣的是,根據回轉半徑和 Dmax 參數,在所有氨酰化結構域突變中,R329H 引發(fā)的構象變化最大(請見Fig. 5B)。
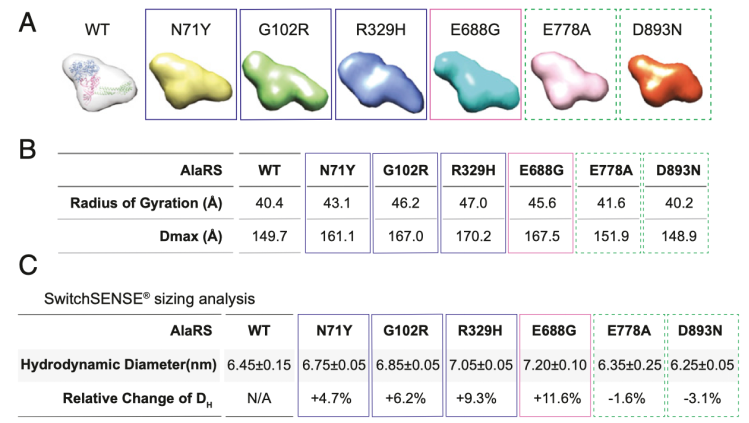
Fig. 5 丙氨酸-核糖核酸酶及其 CMT 突變體的構象分析
氨酰化和校準域突變展示了
更大的流體動力學半徑
我們也使用了switchSENSE技術評估了AlaRS蛋白的流體動力學半徑。這種方法是將短的雙鏈DNA一端固定在金屬表面,另一端以單鏈DNA的形式結合熒光探針,另一條單鏈結合感興趣的蛋白。DNA由芯片表面的電極驅動而上下擺動。一旦DNA鏈和蛋白偶聯(lián),DNA雙鏈的流體動力學摩擦系數受到影響,進而影響DNA納米桿的運動,該運動速度可以用于評估蛋白流體動力學半徑。再次,我們發(fā)現除了C-Ala域的突變,所有的CMT突變,都比野生型的AlaRS具有更大的半徑(請見Fig. 5C)。
只有氨酰化域的突變誘導了
蛋白與Nrp1的異常互作
先前的研究表明,在tRNA合成酶中CMT突變誘導的結構開放性能使得在翻譯機制外發(fā)生異常的相互作用,有助于疾病的發(fā)生。例如,在GlyRS中CMT突變導致了合成酶和跨膜受體Nrp1異常的相互作用。有趣的是,雖然野生型的AlaRS不能與Nrp1發(fā)生互作,三個在氨酰化域的突變能夠誘導蛋白與受體發(fā)生異常的互作(Fig. 6A)。然而,不僅C-Ala域突變而且校準域突變都不能誘導與Nrp1的相互作用(請見Fig. 6A)。使用病人來源的淋巴細胞和正常個體來源的細胞相比,我們確認了攜帶R329H突變的病人中存在AlaRS和Nrp1的相互作用(請見Fig. 6B和C)。
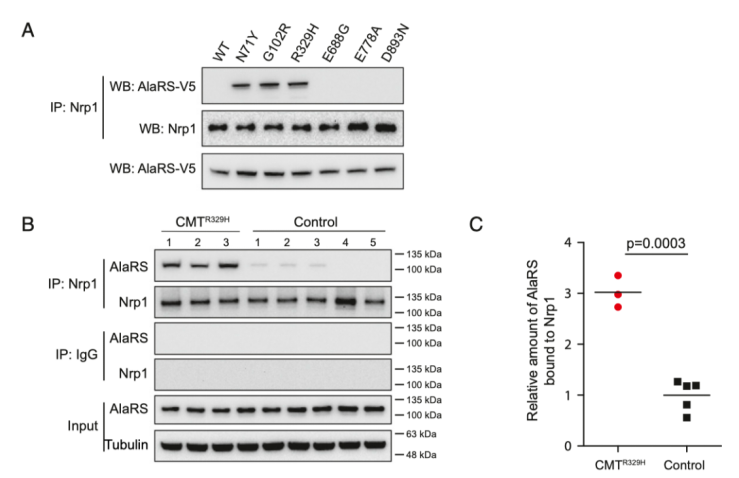
Fig. 6 AlaRS-Nrp1的相互作用

helix+分子互作分析系統(tǒng)采用switchSENSE®技術,通過共價偶聯(lián)或標簽捕獲方式將感興趣的分子(配體)固定在heliX®芯片上,結合標準的自動化工作流程為分子互作提供高效解決方案。
同騰睿杰(上海)生物科技有限公司作為Bruker Dynamic Biosensors中國總代理商,為您提供優(yōu)質的售前售后服務。
聯(lián)系電話:021-50826962
聯(lián)系郵箱:sales@ttbiotech.com

