
研究表明,大部份鈣粘素蛋白家族與疾病,如腫瘤的發(fā)生相關(guān)。由于細胞之間的黏附需要鈣粘素蛋白同源二聚化,相應(yīng)的,能夠調(diào)節(jié)二聚化過程的小分子,將可能成為潛在的治療性藥物。因此,本文介紹了P-鈣粘素特異性小分子的鑒定,作用于P-鈣粘素蛋白介導(dǎo)的細胞黏附過程。雖然該小分子是一種片段化的化合物,但經(jīng)實驗證明,它可以結(jié)合到P-鈣粘素蛋白的凹陷區(qū),此前該位點還未做為藥物靶點。該分子片段能夠阻止一種被稱為X二聚物的中間體形成中的關(guān)鍵氫鍵的形成,進而調(diào)節(jié)了X二聚化的過程。我們的發(fā)現(xiàn)將對小分子用于蛋白-蛋白之間的相互作用調(diào)節(jié)以及蛋白復(fù)合物組裝策略產(chǎn)生重要的影響。
蛋白的組裝揭示了幾乎所有生物學(xué)過程:1. 已有大量被報道的各種功能復(fù)合物的同源或異源結(jié)構(gòu)信息;2. 能夠?qū)Φ鞍捉M裝進行調(diào)節(jié)的調(diào)節(jié)分子可以做為潛在的藥物或探針,用于進一步研究蛋白的功能。但小分子在調(diào)節(jié)蛋白-蛋白相互作用及組裝方面存在困難,部分原因如下:首先,蛋白互作通常會牽涉到較大的分子區(qū)域,但是,小分子可以進入的區(qū)域較小。除此之外,在蛋白-蛋白互作界面,通常不存在明顯的凹槽;缺少能夠指導(dǎo)配體分子設(shè)計的天然蛋白互作分子抑制劑存在。細胞通過鈣粘素蛋白家族,一類鈣依賴性的細胞黏附蛋白分子,發(fā)生細胞之間的黏附,蛋白的組裝。根據(jù)不同的生物組織環(huán)境,鈣粘素分子在各種組織的腫瘤發(fā)生中發(fā)揮作用。P-鈣粘素分子是一種經(jīng)典的鈣粘蛋白,已發(fā)現(xiàn)在許多腫瘤組織中過表達,促進了腫瘤的轉(zhuǎn)移和增殖。細胞之間的聚集體形成,能夠阻止anoikis和P-鈣粘素蛋白介導(dǎo)的信號通路,這對于腫瘤細胞的存活至關(guān)重要。因此,P-鈣粘素蛋白功能的抑制已成為一種潛在的抗癌策略。細胞黏附可以通過不同的細胞間的鈣粘素蛋白反式同源二聚化實現(xiàn),也可以通過相同細胞間的鈣粘素蛋白的順式聚集而成。對于一些類型I的經(jīng)典的鈣粘素蛋白,包括P-鈣粘素,反式同源二聚化已經(jīng)被廣泛的研究。蛋白五個結(jié)構(gòu)域中的兩個細胞外結(jié)構(gòu)域(EC12)經(jīng)證實在蛋白互作的過程中發(fā)揮作用(請見Fig. 1a)。X二聚物,也被稱為鏈-插入二聚物(S-S dimer),是一種中間過渡狀態(tài),能夠促進最終二聚體的形成。S-S二聚物具有一種特定的結(jié)合模式,在該結(jié)合模式之下,一個單體N末端的色氨酸殘基可以插入另一個單體的疏水口袋。根據(jù)這種模式設(shè)計的一些肽類分子模擬配體已有相關(guān)報道,但還未有相關(guān)的配體分子被鑒定,可能是由于這個疏水口袋在S-S二聚化狀態(tài)期間,一直被單體自身或另一個單體的色氨酸殘基所占據(jù),進而影響了對相關(guān)分子機制的更進一步深入研究。在體外進行S-S二聚化過程的抑制研究也許不是最好的方法,我們需要考慮其他方式,研究小分子在調(diào)節(jié)細胞黏附分子組裝方面的功能。
本研究中,我們對這個過程下的任何狀態(tài)進行了小分子篩選,該篩選文庫包括類似藥物的小分片段,鑒定出了一種全新的可以特異性與P-鈣粘素蛋白結(jié)合的小分子。該化合物分子能夠通過調(diào)節(jié)X二聚化抑制細胞之間的黏附,間接的抑制了能夠穩(wěn)定X二聚物的疏水鍵的形成。
該化合物文庫包含1973種化合物。做為初步的篩選,進行了分子之間相互作用檢測。P-鈣粘素蛋白的單體突變蛋白稱為REC12,其中精氨酸被插入到EC12的氨基端,通過生物素-鏈霉親和素捕獲法,被固定在生物傳感器芯片表面,含有100uM化合物的溶液流經(jīng)生物芯片表面。根據(jù)固定的分子水平,REC和化合物的分子量,推測結(jié)合應(yīng)答的RMAX在20RU左右。在1973種化合物文庫中。144種化合物的結(jié)合應(yīng)答效率超過10RU。
作為更進一步的篩選過程,我們將一種稱為EC12的蛋白結(jié)構(gòu)狀態(tài)固定在CM5傳感器芯片表面。這種結(jié)構(gòu)是位于單體和S-S二聚體之間的一種平衡態(tài)。我們進行了一種稱為ABA的檢測實驗,即A和B兩種溶液都流經(jīng)芯片表面。2uM的EC12被稱為溶液A,含有上述第一輪篩選后的化合物的溶液為溶液B,濃度為100uM.對于A溶液,單體結(jié)合應(yīng)答以及單體和芯片之間的共價結(jié)合應(yīng)答,都被檢測到;對于B溶液,兩種化合物明顯的抑制了結(jié)合應(yīng)答效率(請見Fig. 1C)。這些化合物破壞了二聚化的形成,使得單體不能共價結(jié)合到芯片表面。但只有其中一種化合物,Hit 1, 顯示出了劑量依賴性結(jié)合應(yīng)答。單體突變的KD值約為916uM(請見Fig. 1e)。
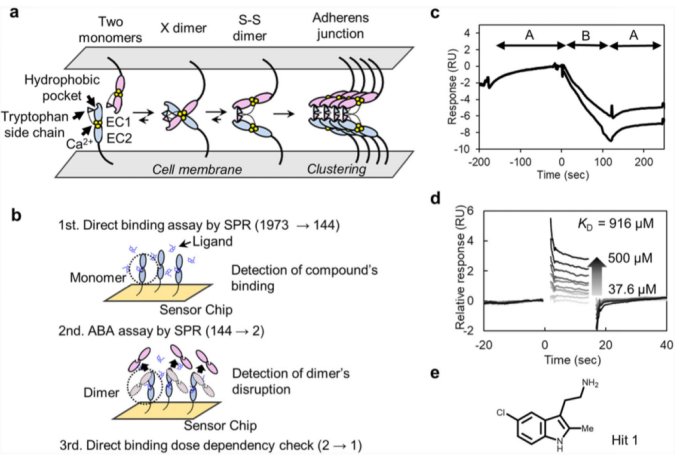
Fig. 1 SPR對潛在化合物篩選示意圖
我們進行了X-射線晶體衍射實驗,用以鑒定Hit 1與單體,REC12的結(jié)合位點,闡明Hit 1的抑制效應(yīng)。REC12經(jīng)結(jié)晶后,與終濃度10mM的Hit 1進行結(jié)合,該復(fù)合物經(jīng)衍射后分辨率為2.30 ? (請見Fig. 2a, Table 1)。位于EC1和EC2之間的結(jié)構(gòu)域的淺腔內(nèi),觀察到了可以用于建模的Hit 1的電子密度分布。Hit 1一經(jīng)與REC12結(jié)合,相對于未結(jié)合Hit 1的REC12,其Y140側(cè)鏈發(fā)生了偏轉(zhuǎn)(請見Fig. 2b),Y140和Hit 1形成了一個CH–π的相互作用。這里需要注意,無論是結(jié)合Hit 1還是未結(jié)合Hit 1的REC12結(jié)構(gòu),都是高度的同型的,因此差異不應(yīng)來自高度同型的Hit 1時,會有一個水分子位于REC12晶體結(jié)構(gòu)的腔內(nèi)。因此,與Hit 1結(jié)合的啟動效應(yīng)有可能來自于和Y140殘基的互作以及脫水作用。同時發(fā)現(xiàn),Hit 1和R68, V98, T99, D100和D137存在范德華力引起的相互作用(請見Fig. 2C)。為了更進一步確認(rèn)在Hit 1結(jié)合后的晶體結(jié)構(gòu)中觀察到的電子密度,我們通過SPR相關(guān)技術(shù),檢測了REC12 Y140R和Hit 1的結(jié)合效應(yīng),發(fā)現(xiàn)Hit 1未能夠和該突變結(jié)合,證實了Y140位點在相互作用中的作用。在結(jié)合口袋周圍的數(shù)個氨基酸殘基,比如Y140,D100和Q101經(jīng)報道對于X二聚化至關(guān)重要。在X二聚體中,可觀測到Y(jié)140和K14,Q101和D100之間存在氫鍵。盡管Hit 1未能直接破壞這些氨基酸殘基之間的氫鍵,但是該區(qū)域結(jié)構(gòu)的改變顯著抑制了X的二聚化。
有趣的是,當(dāng)我們將P-鈣粘素-Hit 1復(fù)合物結(jié)構(gòu)與E-鈣粘素結(jié)構(gòu)進行疊加時,我們發(fā)現(xiàn)E-鈣粘素的腔室體積小于P-鈣粘素的腔室體積。通過CASTp 30對P-鈣粘素,E-鈣粘素結(jié)構(gòu)和P-鈣粘素單體-Hit 1復(fù)合物結(jié)構(gòu)的腔室體積進行測量,也證明了這一點,分別是12.2 ? 3 , 5.4 ? 3 , 21.4 ? 3。該結(jié)果表明較之于E-鈣粘素,P-鈣粘素的腔室更適合于結(jié)合Hit 1分子。此外,F(xiàn)TPap分析得到的31個結(jié)合區(qū)域,都存在于P-鈣粘素中。SPR數(shù)據(jù)同樣表明,Hit 1不與E-鈣粘素結(jié)合,但是能夠與E-鈣粘素N140Y突變體結(jié)合(請見Fig. 2e)。因此,與Hit 1的結(jié)合腔室,能夠做為P-鈣粘素選擇性配體設(shè)計的起始點。
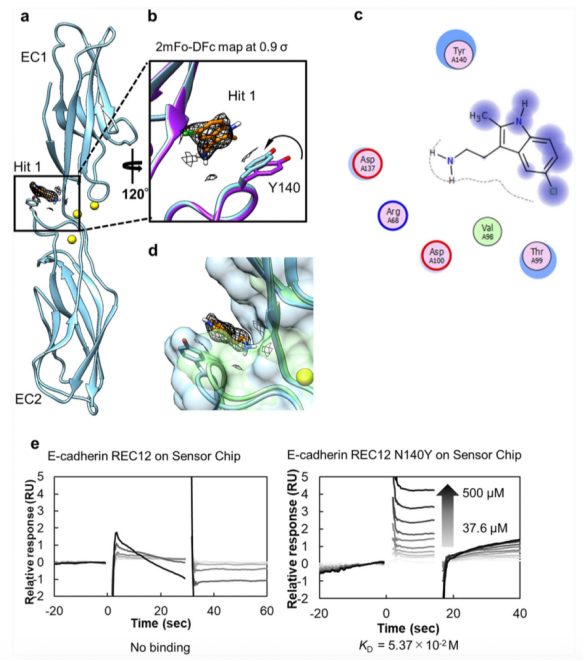
Fig. 2 Hit 1與P-鈣粘素蛋白結(jié)合位點鑒定
為了檢測Hit 1對X二聚化的影響,本文利用了氫-氘交換質(zhì)譜分析法(HDX-MS)。該方法能夠檢測在Hit 1存在與否的情況下,X二聚物的互作面有多大程度暴露在溶劑中。在采用 LC-MS 進行檢測之前先進行胃蛋白酶處理,從而能夠從 P-鈣黏蛋白分子的幾乎所有區(qū)域分離出肽片段。在Hit 1存在與否的情況下,檢測每一段肽段的HDX比率(氫氘交換比率)。在相互作用界面之一(殘基134-140和137-147)的相應(yīng)區(qū)域,該區(qū)域由于在EC12的N端插入了甲硫氨酸,能夠形成X二聚物,但不能形成S-S二聚物,因此抑制了strand-swapping,Hit 1存在時的氫氘交換比率高于未結(jié)合Hit 1時(請見Fig. 3a)。該結(jié)果表明,Hit 1對X二聚物的互作界面能夠產(chǎn)生影響。
為了進一步研究Hit 1如何影響X二聚化,X二聚物(MEC12)經(jīng)結(jié)晶后,與終濃度10mM的Hit 1進行結(jié)合,形成的復(fù)合物結(jié)構(gòu)的分辨率為2.45 ?(請見Fig. 3b)。與上述發(fā)現(xiàn)Hit 1的過程中相同,能夠檢測到三個獨立的電子密度分布,其中的兩個位于Y140殘基周圍的腔室,如Fig. 2a中所示。另一個位于EC2結(jié)構(gòu)域的交匯處(請見Fig. 3b)。位于腔內(nèi)的這些 Hit 1 分子與 Y140 形成了π-π相互作用,并與口袋周圍的殘基或水分子形成了氫鍵連接。由于Hit 1分子能夠與X二聚物的腔室結(jié)合,與口袋周圍的殘基形成多種非共價相互作用,Hit 1和X二聚物結(jié)合的親和性似乎更高。該推測經(jīng)switchSENSE技術(shù)進行了確認(rèn)。Hit 1分子與EC2結(jié)構(gòu)域交匯處的互作主要產(chǎn)生于范德華力。X二聚化中的Y140周圍的腔室結(jié)構(gòu)與單體的不同,Hit 1的結(jié)合模式較單體也有可能存在差異。
與X二聚物無輔因子的狀態(tài)相比,與Hit 1結(jié)合的X二聚物呈現(xiàn)出較顯著的結(jié)構(gòu)變化。EC1-EC2結(jié)構(gòu)域角度變得平滑,B鏈相對于A鏈發(fā)生了轉(zhuǎn)變(請見Fig. 3c)。該角度的結(jié)構(gòu)變化推測可能是由于兩個Hit 1分子結(jié)合在Y140周圍的腔室引起。在 X 多聚體的界面處,K14 的側(cè)鏈與 A138 的主鏈之間形成的氫鍵(即在 Hit 1 結(jié)合時形成的那一個)被破壞了(請見Fig. 3d),可能導(dǎo)致了兩個單體分子的位置轉(zhuǎn)變。
為了進一步確認(rèn)K14側(cè)鏈和A138 的主鏈之間形成的氫鍵對X二聚化形成的重要性,本文使用了尺寸排阻色譜 - 多角度光散射,分別測量了X二聚物(MEC12)和X二聚物突變體K14A的分子量。得到的分子量大小分別是X二聚物為33.8kDa, X二聚物K14A突變體為24.2kDa。
由于在Hit 1結(jié)合的X二聚物中,K14和側(cè)鏈A138之間的關(guān)鍵互作被破壞,我們推測X二聚物的結(jié)構(gòu)可能代表著X二聚化形成過程中的一種不穩(wěn)定狀態(tài)。為了檢測無輔因子存在時的X二聚物的不穩(wěn)狀態(tài)或溶液狀態(tài)下的動力學(xué),我們進行了分子動力學(xué)的模擬。進行了三次各持續(xù) 100 納秒的獨立模擬,并通過 Cα 原子的均方根偏差(RMSD)來確認(rèn)軌跡的收斂性。由此我們計分別計算出了K14A-A138之間我們分別計算的供體氫鍵和受體氧分子之間的距離(請見Fig. 3f),數(shù)據(jù)表明在頻率分布上存在兩個峰,一個是 2.6 ?,另一個是4.8 ?。這種非對稱特性在所有的軌跡中被檢測到。在Hit 1分子結(jié)合下的蛋白晶體結(jié)構(gòu)中,距離則分別為 4.6 和2.6 ?。上述結(jié)果表明,在氫鍵形成時,無輔因子狀態(tài)下的X二聚物動力學(xué)可能存在兩種狀態(tài),與Hit 1結(jié)合狀態(tài)下的X二聚物的兩種晶體動力學(xué)狀態(tài)一致。換句話說,apo狀態(tài)的 X 大分子能夠呈現(xiàn)出與在晶體結(jié)構(gòu)中與 Hit 1 結(jié)合的 X 大分子所呈現(xiàn)的構(gòu)象非常接近的構(gòu)象,即便沒有 Hit 1 化合物的存在也是如此。表明,在晶體結(jié)構(gòu)中觀測到的Hit 1結(jié)合下的X二聚物的狀態(tài)是X二聚化過程中的一種狀態(tài)。總的說來,Hit 1的結(jié)合可能會通過將X二聚物限定在二聚化過程的一種狀態(tài)下,進而干擾對X二聚化非常重要的氫鍵的形成。和orthosteric抑制劑不同,Hit 1不會直接的阻止兩個單體之間氫鍵的形成,而是可能通過改變單體結(jié)構(gòu),進而阻止氫鍵的形成。
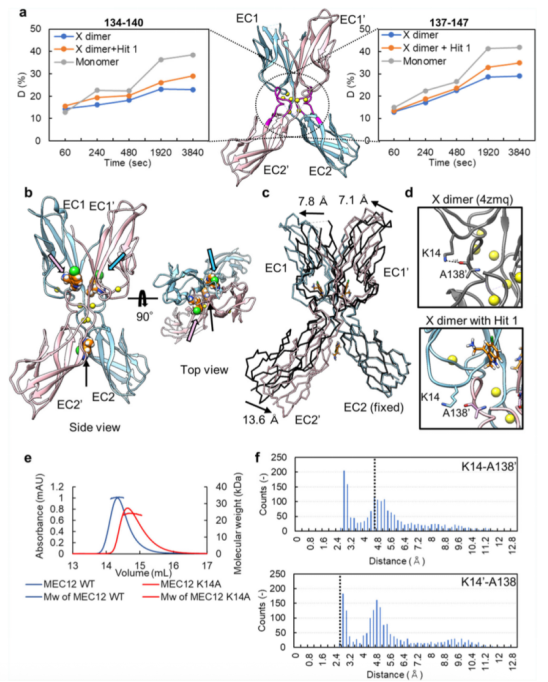
Fig. 3 Hit 1對蛋白二聚化的影響鑒定
我們進一步通過細胞-聚集檢測技術(shù),驗證了Hit 1是否能夠抑制細胞之間的黏附作用。我們通過Flp-In-CHO體系,構(gòu)建了一種表達P-鈣粘素的CHO細胞系。在該檢測中,除了P-鈣粘素蛋白,細胞外蛋白經(jīng)胰蛋白酶消化,使得細胞之間的黏附和細胞聚集體的形成僅僅取決于P鈣粘素分子的相互作用。實驗表明,空白對照組CHO細胞之間未能夠形成大的細胞聚集體(請見Fig. 4a,c)。在胰蛋白酶消化后,培養(yǎng)基替換成不含鈣離子的培養(yǎng)基,以防止細胞聚集,通過添加1mM的CaCl2促進細胞聚集。在細胞發(fā)生聚集反應(yīng)后,使用微流控成像(MFI)設(shè)備測量細胞聚集體尺寸大小分布。在MFI的測量中,大于40um的細胞聚集體被定義為P-鈣粘素依賴的細胞聚集,因為在EDTA存在時,會有大量的大小在25-40um的細胞聚集體存在。為了對數(shù)量進行標(biāo)準(zhǔn)化,通過計算大于40um-100um的顆粒數(shù)與大于25-100um的顆粒數(shù)的比值實現(xiàn)。在對照實驗中,EDTA能夠抑制大的聚集體(40-100um)的形成,因此細胞黏附取決于鈣依賴性的P-鈣粘素分子的相互作用(請見Fig. 4a,c)。
當(dāng)同時添加Hit 1和1mM的CaCl 2分子時,聚集體的形成呈現(xiàn)Hit 1濃度依賴的方式(請見Fig. 4b, c)。而當(dāng)Hit 1加入到已經(jīng)形成聚集體的培養(yǎng)體系中時,在檢測時間段,形成的聚集體仍舊穩(wěn)定存在,而EDTA能夠部分的破壞已經(jīng)形成的聚集體。這些結(jié)果說明,Hit 1是通過阻止X二聚化形成過程中兩個單體分子的互作,一旦細胞聚集體形成,Hit 1是無法破壞它們的互作穩(wěn)定性的。
為了對Hit 1在X二聚化過程中的效應(yīng)進行量化分析,我們進行了脂質(zhì)體聚集檢測。我們首先制備了帶有 C 端組氨酸標(biāo)簽的 X 二聚體(MEC12),然后將該蛋白質(zhì)與一種基于 DOPC 的脂質(zhì)體一起孵育,該脂質(zhì)體中含有 10% 的具有鎳吸附作用的脂質(zhì) DOGs-NTA-Ni,并在 EDTA 存在下使 P-鈣黏蛋白分子失活。在這些條件之下,在650nm的吸收光下,未觀測到聚集(請見Fig. 4d)。我們也同時觀測到,脂質(zhì)的聚集僅僅在MEC12和 CaCl 2都存在時發(fā)生(請見Fig. 4d)。一旦CaCl 2添加到溶液中,脂質(zhì)體聚集,推測是通過被捕獲在脂質(zhì)體表面的C端帶有組氨酸標(biāo)簽的X二聚物(MEC12)的二聚化引起,可通過逐漸增加的650nm處的吸光值檢測,該吸光值最后可達到平臺期(請見Fig. 4d)。通過使用一相結(jié)合的指數(shù)衰減方程模型,計算出了脂質(zhì)體聚集的觀測速率常數(shù)(kobs),以此作為 X 雙聚體化動力學(xué)的指標(biāo)。Hit 1存在時的速率常數(shù)(k obs = 0.0033) 顯著低于Hit 1未存在時的速率常數(shù) (k obs = 0.0086)(請見Fig. 4e)。此處需注意,這并非是由于鈣粘分子的激活引起,因為通過DSC測量可知,無論Hit 1存在與否,鈣離子與鈣粘素分子的結(jié)合并未被Hit 1所抑制。該結(jié)果表明,Hit 1通過間接的破壞對X二聚化形成關(guān)鍵的氫鍵,抑制了X二聚化的過程,進而抑制了細胞的聚集。
我們利用兩種細胞系分析了Hit 1對于內(nèi)源性P-鈣粘素分子介導(dǎo)的細胞黏附的效應(yīng),以及對于不表達P-鈣粘素分子的細胞聚集效應(yīng)。其中HCT116表達內(nèi)源性P-鈣粘素分子,MCF7細胞不表達P-鈣粘素分子。將細胞種植在培養(yǎng)板中,并添加100uM的Hit 1分子,細胞聚集程度通過對整體區(qū)域面積下的聚集體進行定量分析。由于HCT116和MCF7的每一個單細胞區(qū)域都不同,標(biāo)準(zhǔn)化后的細胞區(qū)域經(jīng)Hit 1存在時,每一個濃度下的整體細胞區(qū)域比上Hit 1不存在時整體細胞區(qū)域計算得到。在100uM的Hit 1存在時,HCT116細胞標(biāo)準(zhǔn)化后的細胞區(qū)域,下降到未加入Hit 1時的細胞區(qū)域的75.7%,但Hit 1對MCF7細胞標(biāo)準(zhǔn)化后的區(qū)域沒有產(chǎn)生影響(請見Fig. 4f)。已有研究報道,P-鈣粘素蛋白敲除后對腫瘤轉(zhuǎn)移具有一定的影響,以及對細胞內(nèi)信號通路的影響。本研究中結(jié)合已有的表型研究表明,在表達P-鈣粘素分子的腫瘤細胞中,Hit 1能夠抑制細胞之間的黏附作用,進而導(dǎo)致β -catenin信號通路的下調(diào)及細胞凋亡信號的激活。
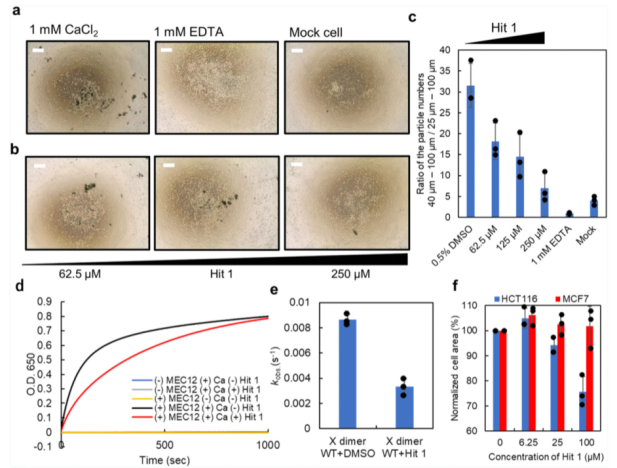
Fig. 4 Hit 1抑制細胞黏附檢測
由于Hit 1以及初篩鑒定的許多化合物都包括陽離子功能基團,我們因此通過基于吲哚的商業(yè)化合物:tryptamine, tryptophan, 和 auxin 鑒定了陽離子功能基團對于結(jié)合活性的重要性(請見Fig. 5a)。令人驚訝的是,只有tryptophan與 P-鈣黏蛋白結(jié)合,這可能是由于Hit 1結(jié)合口袋周圍蛋白表面存在著負電荷。該結(jié)果表明,在進一步的藥物發(fā)現(xiàn)中,不應(yīng)對陽離子基團進行修飾改造。根據(jù)Hit 1和P-鈣粘素蛋白的2D相互作用圖譜的研究(請見Fig. 2c),我們制定了兩個進一步合成的策略:(1)將吲哚環(huán)第二個位置的甲基替換成羧基,這將導(dǎo)致羧基和R68,氨基和D137之間形成鹽橋;(2)將吲哚環(huán)中 C5 位置的氯原子替換為一種更大分子量的官能團,抑制第二個單體的接近。我們首先合成了3-(2-氨基乙基)-5-苯基-1H-吲哚-2-羧酸(簡稱 Hit 1-羧酸,請見Fig. 5b),但是從SPR親和性檢測來看,它與REC12的親和性并不優(yōu)于Hit 1,也不具備細胞聚集抑制效應(yīng)。接下來,我們合成了 2-(2-甲基-5-苯基-1H-吲哚-3-基)乙烷-1-胺(苯基-Hit 1)(請見Fig. 5b)。通過SPR互作檢測分析,我們觀測到一種劑量依賴性應(yīng)答;其與REC結(jié)合的親和性與Hit 1相同。在脂質(zhì)體聚集檢測中,苯基-Hit 1相比較于Hit 1,具有更強的抑制活性。tryptamine, auxin 和 tryptophan對聚集效應(yīng)影響較小(請見Fig. 5c)。Tryptamine 不具有大的功能基團,這可能是它不能抑制脂質(zhì)體聚集的原因。在細胞聚集檢測中,苯基-Hit 1相較于Hit 1具有更強的抑制效應(yīng),且苯基-Hit 1與X二聚體的結(jié)合親和性要強于單體P-鈣粘素蛋白。
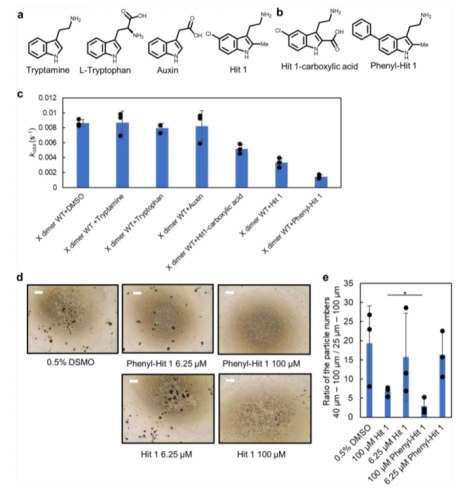
Fig. 5 抑制劑設(shè)計策略

