
在腫瘤研究和精準醫療領域,腫瘤類器官(如腫瘤球體)作為一種高度仿生的體外模型,正逐漸成為基礎研究、藥物開發和個性化醫療的重要工具。然而,傳統的腫瘤球體培養方法存在諸多局限性,例如操作復雜、成本高昂、細胞需求量大以及難以實現高通通量篩選等。本篇提出了一種基于滴液微陣列(Droplet Microarray)平臺的創新方法,結合I.DOT非接觸式移液技術,實現了腫瘤球體的高效培養和藥物篩選。
腫腫瘤球體是由癌細胞自發聚集形成的三維球狀結構,能夠高度模擬體內腫瘤的微環境[1]。與傳統的二維細胞培養相比,腫瘤球體具有以下特點:
腫瘤球體能夠自主合成ECM蛋白,這些蛋白與細胞間的接觸共同賦予了球體獨特的力學特性和生物學行為[2]。
這種特殊的微環境與體內腫瘤相似,能夠誘導癌細胞的侵襲性、增殖能力和藥物耐受性[3-5]。
在腫瘤球體中進行的藥物敏感性和耐受性測試,其結果與體內實驗更為接近[6-8]。
然而,傳統的腫瘤球體培養方法存在諸多不足,例如細胞數量需求大、培養體積大、難以實現單球體培養等。這些問題限制了其在高通量藥物篩選中的應用。
I.DOT與滴液微陣列:
腫瘤球體培養的新突破
本研究利用I.DOT非接觸式移液技術結合滴液微陣列(DMA)平臺,開發了一種高度微型化、自動化的腫瘤球體培養和篩選系統[9-10]。該系統的核心優勢包括:
通過在納升至微升范圍內進行液體分配,I.DOT技術顯著減少了培養基、試劑和細胞的消耗。
DMA平臺能夠在超疏水背景上形成均勻的親水點陣列,每個液滴中僅培養一個腫瘤球體,避免了傳統方法中多個球體共存導致的實驗誤差。
該系統能夠在短時間內形成大量均勻的腫瘤球體,并直接在平臺上進行藥物處理和顯微鏡分析。
DMA平臺由親水點陣列組成,背景為超疏水表面,能夠形成均勻、獨立的納升液滴。使用兩種類型的DMA滑片:一種涂覆有羥乙基甲基丙烯酸酯(HEMA)-乙二醇二甲基丙烯酸酯(EDMA),另一種未涂覆聚合物層。滑片的表面由超疏水背景上的親水性斑點陣列構成,親水性斑點的水接觸角(WCA)≤10°,滑動角(SA)>90°,超疏水邊界的WCA=154±1.5°,SA=2.4±0.5°。這種特殊的設計使得在其表面滴加水溶液時,能自發形成高密度、均一、分離且穩定的納米液滴陣列。每個液滴都可以捕獲細胞,成為進行個體生物學實驗的“小反應器”(圖1&圖2)。
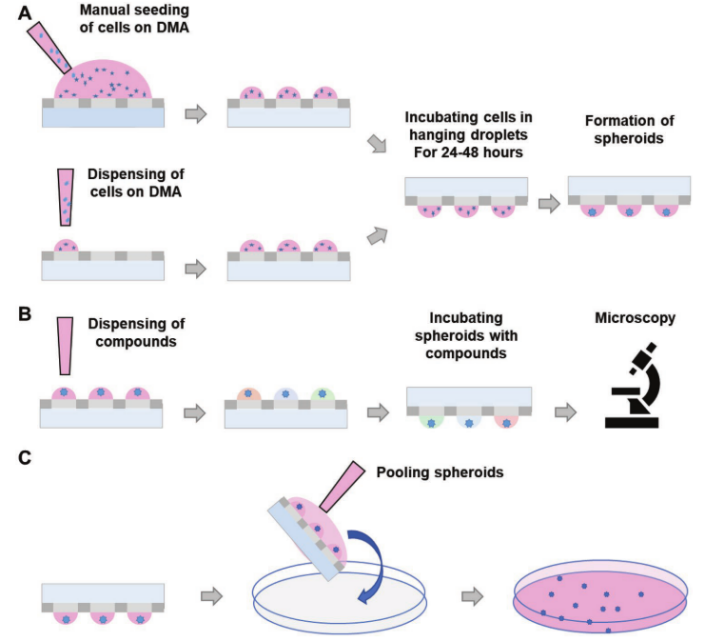
圖1. 示意圖展示了在液滴微陣列(DMA)平臺上制造和篩選球狀體。a)手動細胞接種和通過分配接種,然后在懸滴中生長球狀體。b)使用單球體微陣列的化合物篩選。c)匯集在DMA平臺上形成的球狀體。
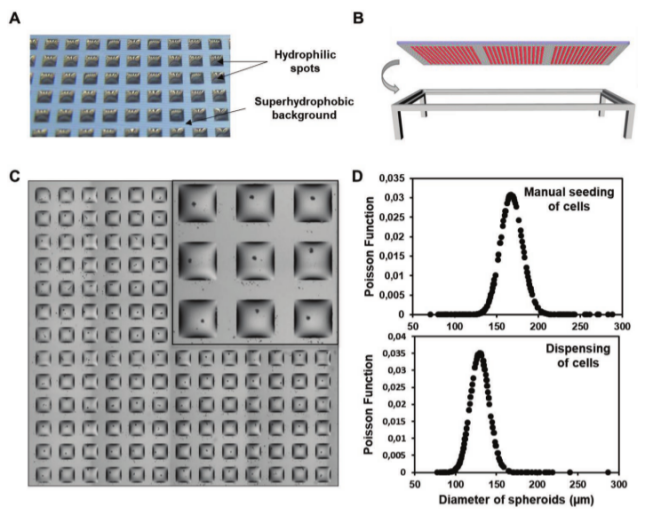
圖2. a)DMA載玻片上形成的水滴陣列的照片。比例尺:1 mm。b)懸滴格式的DMA載玻片。c)含有單個球狀體的13 × 13液滴陣列的顯微鏡圖像。比例尺:1mm。插圖:0.5mm。d)顯示球狀體直徑分布的圖。通過使用手動接種(上圖)和通過細胞分配(下圖)獲得球狀體。利用Poisson函數在Excel程序中繪制球體尺寸分布圖,每張圖分析196個球體。
研究人員從多種癌細胞系(包括HeLa、MCF-7和HEK293)中培養腫瘤球體(圖3)。細胞首先被稀釋至特定濃度(手動接種為2.5×105 cells/mL,自動化接種為1.5×106 cells/mL),然后通過I.DOT非接觸式移液技術將細胞以100納升液滴的形式分配到DMA平臺上,每個液滴中包含約150個細胞。隨后,DMA滑片被倒置,形成懸掛液滴,細胞在液滴中自發聚集形成球體。在不更換培養基的情況下養數天,監測球狀體的生長和活力(圖3a,b,e,f)。結果顯示,球體大小可通過改變初始接種細胞濃度來控制,而且大多數球體形狀規則,邊緣光滑。在不更換培養基的情況下培養,HeLa球體直徑在24-96小時內從70±10μm增長到100±15μm(圖3a,b),HEK293和MCF-7球體直徑有所減小,但三種細胞系球體在培養7天內活力均高于90%(圖3e,f)。
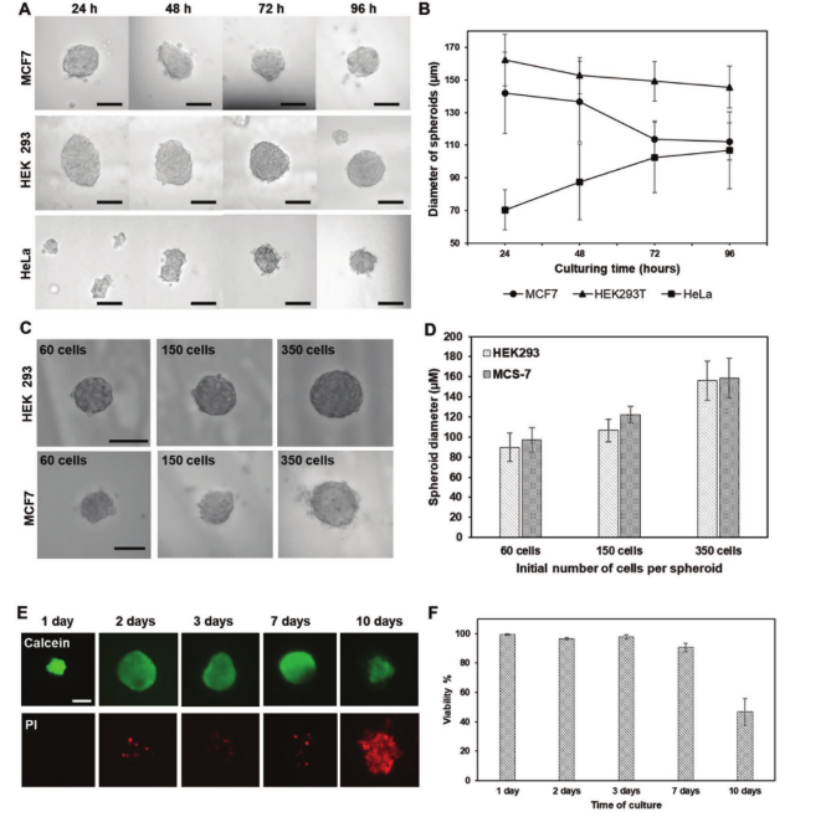
圖3. 在微滴微陣列(DMA)平臺上制造和培養球狀體。a)在細胞接種后24、48、72和96小時,由MCF-7、HEK 293和HeLa細胞形成的球狀體的雙視野顯微鏡圖像。比例尺:100 μm。b)從在80 nL液滴中培養96小時的MCF-7、HEK 293和HeLa細胞系獲得的球狀體的直徑。初始細胞數平均為150個細胞/球狀體。c)由HEK 293和MCF-7細胞系形成的球狀體的雙視野顯微鏡圖像,從約60、150和350個細胞/球狀體開始。比例尺:100 μm。d)從不同初始細胞數開始的HEK 293和MCF-7細胞系獲得的球狀體的直徑。e)在DMA上培養1、2、3、7和10天而不更換培養基的HEK 293球狀體的顯微鏡圖像。用鈣黃綠素AM和碘化丙啶(PI)染色球體。比例尺:100 μm。f)由在DMA上培養10天而不更換培養基的HEK 293細胞形成的球狀體的活力。* 所有圖表上的誤差線表示標準偏差。
用三種抗癌化合物(阿霉素、奧沙利鉑和5-氟尿嘧啶)對HeLa細胞形成的腫瘤球體進行處理,并與2D單層培養的HeLa細胞進行劑量-反應對比(圖4)。HeLa細胞以150個細胞/100 nL液滴的密度接種,培養48小時形成球體后進行藥物處理;2D培養的HeLa細胞接種后先培養5小時使其貼壁,再進行藥物處理。藥物處理72小時后,用鈣黃綠素和碘化丙啶(PI)染色觀察活細胞和死細胞。
結果顯示,三種化合物對3D和2D培養的HeLa細胞活力均有明顯的劑量依賴性影響(圖4),但腫瘤球體中的HeLa細胞對藥物治療的耐藥性更強。例如,阿霉素對3D培養的HeLa細胞的IC50約為6 μM,對2D培養的HeLa細胞的IC50<0.05×10-6M。
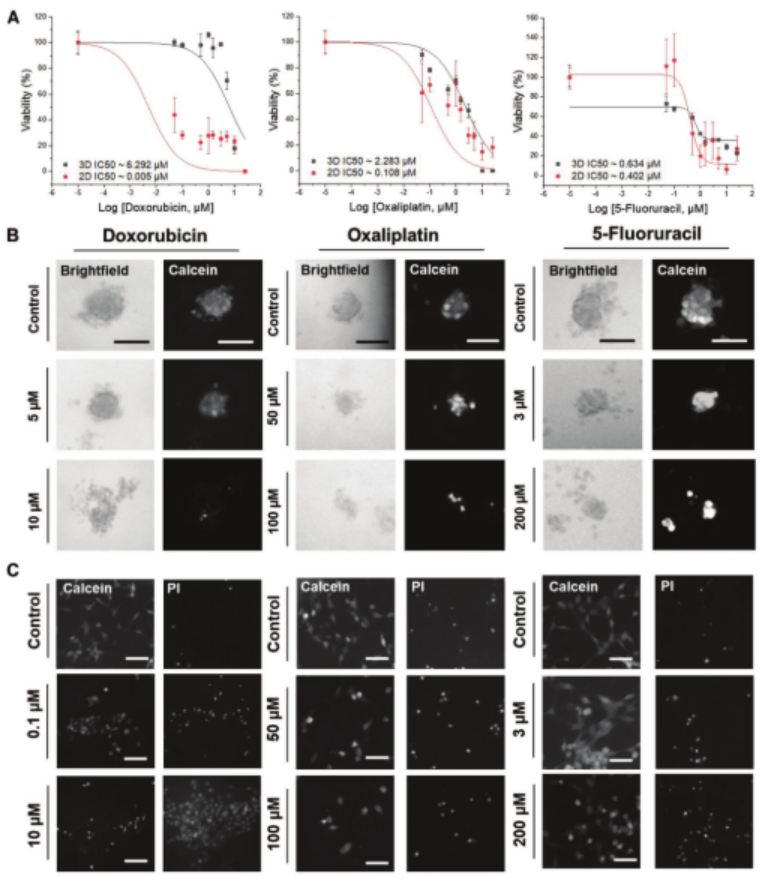
圖4. 用抗癌藥物在液滴微陣列(DMA)平臺上以3D球狀體和2D單層培養的HeLa細胞的治療。a)多柔比星、奧沙利鉑和5-氟尿嘧啶對以3D和2D形式培養的HeLa細胞的劑量依賴性作用的比較。初始細胞數:對于2D和3D細胞培養物,每個斑點約150個細胞。球狀體形成72小時,然后使用非接觸式液體分配器添加藥物,并在染色和顯微鏡分析之前將球狀體孵育48小時。使用“非線性曲線擬合”功能在OriginPro中繪制劑量-反應曲線。曲線擬合后,在OriginPro中計算IC 50值。B)用不同濃度的藥物處理的HeLa球狀體的代表性顯微鏡圖像。比例尺:100 μm。c)用不同濃度的藥物處理的在2D單層中培養的HeLa細胞的代表性顯微鏡圖像。比例尺:100 μm。
液滴微陣列平臺在腫瘤球體研究方面具有顯著優勢。與傳統微孔板相比,它的液滴體積可縮小至100 nL,能節省高達99%的化合物、試劑和細胞,大幅降低成本,這為藥物研發早期的超高通量篩選提供了可能。而且,每個球體僅需150個細胞,對于稀有且難以擴增的細胞,如原發性患者來源的腫瘤細胞的研究至關重要。
該平臺還可以通過改變初始細胞數量和斑點大小來培養不同大小的球體,不同大小的球體對藥物治療的反應可能不同,能滿足不同研究需求。另外,在DMA玻片上可直接對球體進行顯微鏡觀察,無需轉移,簡化了操作流程。形成的均一細胞球體還能方便地收集用于后續實驗,在體外測試到再生醫學等領域都有重要應用。
這項研究展示了液滴微陣列平臺在構建和篩選腫瘤球體方面的巨大潛力,有望推動基礎癌癥研究、藥物研發和精準醫學的發展。期待未來該平臺能在更多領域發揮重要作用,為攻克癌癥帶來新的希望。

該同騰睿杰(上海)生物技有限公司作為CYTENA I.DOT中國總代理商,為您提供優質的售前售后服務。
聯系電話:021-50826962
聯系郵箱:sales@ttbiotech.com
1. B. Mayer, A. Tischer, A. Wieser, K.-W. Jauch, I. Funke, Mol. CancerTher. 2007, 6, A215.
2. S. A. Langhans, Front. Pharmacol. 2018, 9, 6.
3. Y. Dai, K. Bae, D. W. Siemann, Int. J. Radiat. Oncol., Biol., Phys. 2011, 81, 521.
4. M. H. Ryu, H. M. Park, J. Chung, C. H. Lee, H. R. Park, Biochem. Biophys. Res. Commun. 2010, 393, 11.
5. N. J. Sullivan, A. K. Sasser, A. E. Axel, F. Vesuna, V. Raman, N. Ramirez, T. M. Oberyszyn, B. M. Hall, Oncogene 2009, 28, 2940.
6. K. M. Yamada, E. Cukierman, Cell 2007, 130, 601.
7. G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker, S. Takayama, J. Controlled Release 2012, 164, 192.
8. M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua, A. Tesei, Sci. Rep. 2016, 6, 19103.
9. W. Q. Feng, L. X. Li, E. Ueda, J. S. Li, S. Heissler, A. Welle, O. Trapp, P. A. Levkin, Adv. Mater. Interfaces 2014, 1, 1400269.
10. A. N. Efremov, E. Stanganello, A. Welle, S. Scholpp, P. A. Levkin, Biomaterials 2013, 34, 1757.

