
單克隆抗體的親和力的篩選及優化對候選藥物的開發至關重要,因為它可以影響藥物的療效,從而影響劑量和給藥方案。本研究聚焦于優化一種創新的“二合一”抗體,該抗體能夠同時靶向表皮生長因子受體(EGFR)和程序性死亡配體1(PD-L1),由于替換了輕鏈CDR3區域的單個氨基酸,分離的抗體變體靶向EGFR的親和力提高了60倍。同時使用多種方法確認了二合一變體的結合特性,包括應用BLI、switchSENSE®及原生分子互作細胞分析系統(RT-ICRT-IC)等方法進行驗證。結合實驗研究團隊通過使用位點定向突變和酵母表面展示技術,成功地對EGFR結合位點進行了親和成熟。該研究不僅展示了抗體工程在提高藥物療效方面的潛力,還為未來的癌癥治療提供了新的思路。
在過去幾十年中,單克隆抗體(mAbs)已成為治療各種疾病的重要藥物類別。為了提高療效并減少副作用,抗體對其抗原具有高親和力和特異性至關重要[1]。抗體的抗原結合位點由6個互補決定區(CDRs)組成,其中3個位于重鏈上,3個位于輕鏈上[2]。在抗體親和力成熟過程中,CDRs經歷了高度的體細胞突變。在哺乳動物中,通過免疫球蛋白(Ig)基因的突變多樣性產生高親和力抗體,這一過程稱為體細胞超突變(SHM)[3,4]。
由于Ig基因的隨機SHM和含有親和力增強突變的B細胞的選擇及克隆擴增之間的交替過程,在免疫反應中抗體的親和力大幅提升[5]。治療性抗體可以來源于多種物種,如小鼠、大鼠、家兔、雞或非人靈長類動物[6,7]。動物免疫的優勢在于抗體已經通過幾輪SHM在體內經歷了親和力成熟[8]。然而,即使是體內衍生的抗體也可能沒有對抗原所需的親和力,這隨后可以通過體外親和力成熟進行優化[9]。為此,可以應用隨機或有針對性的突變[8]。
靶向誘變方法通過定點誘變產生抗體變體庫,突變可設計CDR區域[8,10]。可通過展示淘選技術(如噬菌體展示[11]、酵母表面展示[12]或核糖體展示[13-14])進行親和篩選來分離最佳成熟的抗體變體。但對于包含所有可能單突變和多突變組合的抗體庫,其規模可能超1030個變體,難以用常規展示技術篩選,而熱點誘變、透視誘變和同步誘變可提高CDR多樣化效率并減小庫的規模[15-17]。
對于單特異性抗體,優化親和力可影響藥物療效、劑量和給藥方案,限制不良反應并降低治療成本。而雙特異性抗體(bsAbs)在癌癥治療中具有獨特優勢,能夠交聯受體或阻斷兩種疾病相關的信號通路,通過同時靶向同一惡性細胞上的兩種癌癥特異性抗原,可增強腫瘤特異性[18-20]。在許多實體瘤中,EGFR和PD-L1是兩種重要的治療靶點 [21,22]。該研究針對一種同時靶向EGFR和PD-L1的Two-in-One抗體(HCP-LCE)展開。這種抗體通過對兩只雞分別用EGFR和PD-L1胞外域免疫,然后將各自的重鏈和輕鏈模塊組合成Fab格式以分離同時結合兩個靶點的抗體變體[23]。HCP-LCE通過結合二聚化結構域II抑制EGFR信號傳導,并阻斷PD-1/PD-L1相互作用,對每個抗原具有中等個體親和力。
該研究旨在探究能否在不喪失對PD-L1結合能力的前提下,優化該抗體對EGFR的親和力,這是首次對動物免疫和組合篩選獲得的Two-in-One抗體進行親和力成熟研究,通過位點定向誘變和酵母表面展示(YSD)結合熒光激活細胞分選(FACS)優化 HCP-LCE對EGFR的親和力。
為構建酵母文庫,該研究設計針對輕鏈CDR1(LCDR1)和CDR3(LCDR3)特定氨基酸的引物,通過PCR反應構建文庫。將VL基因通過同源重組轉入酵母載體,與HCP-LCE的重鏈結合進行Fab展示。誘導基因表達后,用不同濃度的EGFR-Fc等進行孵育,再用相應抗體檢測,最后通過FACS篩選。
對篩選出的酵母載體測序,將VL片段重新格式化到pTT5衍生載體,轉染Expi293FTM細胞表達全長抗體,收集細胞培養上清,用MabSelectTM PrismA HP柱純化,再進行緩沖液交換。
Expi293FTM細胞在Expi293TM表達培養基中培養,每3-4天傳代一次,37℃、8% CO2孵育。A431和A549細胞在T75細胞培養瓶中,在37℃和5% CO2條件下,在Dulbecco 's Eagle培養基中添加10% FBS和1%青霉素-鏈霉素中培養,每3-4天傳代一次。Jurkat細胞保存在添加10% FBS和1%青霉素-鏈霉素的RPMI-1640中,每3-4天傳代一次,37℃和5% CO2孵育。
在CFX Connect Real-Time PCR Detection System中,設置溫度梯度,測定抗體變體在不同溫度下的穩定性,計算熔解溫度(Tm)。
使用顯示EGFR-ECD截斷版本的酵母細胞在亞結構域水平上進行EGFR表位定位。與相應抗體孵育,用FITC共軛抗-c-myc抗體和抗人IgG Fc PE共軛抗體分別驗證表面展示和抗體結合,通過流式細胞術(CytoFLEX S系統)分析細胞。
0
6用Octet RED96系統,將HCP-LCE變體固定在抗人IgG-Fc捕獲生物傳感器上,分別與不同濃度的EGFR-ECD和PD-L1-ECD孵育,測定親和力和結合動力學參數。用ForteBio數據分析軟件9.0.0.14分析數據,采用Savitzky-Golay過濾和1:1 Langmuir結合模型確定結合動力學參數。
在PD-1競爭實驗中,將HCP-LCE和LCE-E固定在抗人Fab-CH1第二代生物傳感器上。為了進行同時結合實驗,AHC生物傳感器上裝載了單臂(oa)版本的HCP-LCE或LCE-E。作為對照,oaHCP-LCE和oaLCE-E僅與PD-L1孵育。
switchSENSE®測量在helix+儀器(Dynamic Biosensors,Munich, Germany)中進行,使用標準適配器芯片(ADP-48-2-0, Dynamic Biosensors)在靜態熒光接近傳感(FPS)模式下進行。在準備測量時,使用heliX®胺偶聯試劑盒1 (HK-NHS-1, Dynamic Biosensors)將目標蛋白偶聯到配體鏈上。蛋白-DNA偶聯物在proFIRE®(Dynamic Biosensors)中純化。兩個蛋白-DNA偶聯物被分離。實驗采用較早的洗脫共軛物(共軛物1)。PD-L1配體鏈偶聯物與適配器鏈1用紅色染料Ra (AS1-Ra, Dynamic Biosensors)混合,EGFR配體鏈偶聯物與適配器鏈1用綠色染料Ga (AS1-Ga, Dynamic Biosensors)混合20 min,溫度25℃。
對于單蛋白表面實驗,20% PD-L1-LS/AS1-Ra或20% EGFR-LS/AS1-Ga與80%互補錨鏈1 (c-Anchor 1, DC-0, Dynamic Biosensors)混合。對于雙蛋白表面實驗,電極1的功能化混合物含有10% PD-L1-LS/AS1-Ra, 10% EGFR-LS/AS1-Ga和80%互補錨鏈1 (c-Anchor 1, DC-0, Dynamic Biosensors)。電極2的功能化混合物包含無配體鏈(LFS-0,Dynamic Biosensors)與具有相應染料(AS-2-Ra和AS-2-Ga,Dynamic Biosensors)的適配器鏈2混合,并以相應比例與c-錨2 (DC-0,Dynamic Biosensors)混合。工作流在heliOS軟件v2024.1.0中設置。表面被功能化了200秒。從10倍濃縮緩沖液(BU-PE-140-10, Dynamic Biosensors)中稀釋1倍PE140,將分析物稀釋至1E-8 M, 1E-9 M和1E-10 M,作為運行緩沖液。以200 μL/min的流速測定180 s的關聯。以500 μL/min的流速測定1800s的解離。在僅PD-L1的實驗中,紅色LED功率設置為2%,綠色LED功率設置為0%。
在EGFR-only實驗中,綠色LED功率設置為2%,紅色LED功率設置為0%。在雙蛋白/雙色實驗中,兩個led都使用2%的功率。測量在25°C下進行。用heliOS軟件v2024.1.0對測得的結合曲線進行評價。所得的雙參考數據點采用連續振幅的單相結合-雙相解離模型進行擬合。使用R package ggplot2繪制擬合曲線。
在將A549細胞與不同濃度的抗體孵育,再用抗人IgG Fc PE共軛抗體檢測,通過流式細胞術測定細胞結合情況,所得曲線采用GraphPad Prism進行參數擬合。
在芯片型號為M5 (CY-M5-1, Dynamic Biosensors)的heliXcyto裝置(Dynamic Biosensors)上測量細胞的實時動力學。抗體用紅色染料標記,使用heliXcyto標記試劑盒red dye 1 (CY-LK-R1-1, Dynamic Biosensors),按照說明書進行標記。用光度法測定標記度(DOL)。HCP-LCE和LCE-E的測定DOL分別為2.5和4。將細胞重懸于PBS中,并通過30 μm細胞濾器(FIL-30-20, Dynamic Biosensors)進行篩選。除了A431細胞上的LCE-E的一次測量外,所有的相互作用都是在室溫下用2% PFA處理15分鐘的細胞上測量的。細胞以2×106個/mL的濃度捕獲。抗體HCP-LCE和LCE-E分別稀釋至60/20/6.67 nM和100/50/25 nM。使用從10倍原液(BU-RB-10-1, Dynamic Biosensors)稀釋的1×PPBS進行抗體稀釋,并作為運行緩沖液。工作流程在heliOS軟件v2024.1.0中設置。用相應濃度的紅色染料(NOR-0, Dynamic Biosensors)的歸一化溶液對信號進行歸一化處理。紅色LED功率設置為0.11-0.13%。測量在25℃下進行,自動進樣器冷卻至4℃。測定每一種結合作用300 s,測定每一種解離作用1800 s。利用heliOS軟件v2024.1.0對測得的結合曲線進行評價。根據歸一化溶液得到的信號,對Spot 1和Spot 2的結合曲線進行歸一化處理。使用R package ggplot2繪制并擬合曲線。
用ColabFold版本1.5.5進行建模[24-27],MMseq2生成多個序列比對(msa)[28,29],使用UniRef100數據庫[30]并通過AMBER力場松弛模型[31],用PRODIGY計算結合能和解離常數[32]。EGFR使用AlphaFold2對接,隨后利用HDOCK對接PD-L1[33]。
通過篩選雞源酵母表面展示文庫確定二合一抗體HCP- LCE的EGFR結合模塊(圖1A),該文庫由抗PD-L1抗體重鏈與免疫抗EGFR輕鏈文庫結合而成。因HCP-LCE同一Fv 區同時靶向EGFR和PD-L1且PD-L1結合主要靠重鏈CDR,所以認為輕鏈CDR主要參與EGFR結合[23]。
為使 HCP-LCE的EGFR結合親和力成熟,隨機化 LCDR1 和 LCDR3 的單氨基酸生成YSD文庫(圖1B),未對LCDR2修飾。YSD文庫設計為LCDR1的9個氨基酸、LCDR3的12個氨基酸各含一個突變,每個輕鏈最大突變率為2個突變,采用位點飽和誘變[34],用退化的NNS密碼子減少終止密碼子(圖1B),組合所有可能突變對,理論文庫大小在蛋白和DNA水平分別為4.3×10?個輕鏈突變體和1.1×10?個突變體。NNS YSD文庫克隆時,VL片段經同源重組插入載體,再通過酵母交配與編碼野生型二合一 VH-CH1片段的酵母細胞結合(圖1B)。
二倍體二合一 LC NNS文庫經 FACS 篩選高親和力EGFR結合物,共三輪(圖1C),從500 nM EGFR-fc開始。第二輪篩選進行競爭篩選,用100 nM 標記的EGFR與500 nM野生型抗體預孵育對富集文庫染色(圖1C),雙陽性酵母細胞表明其靶向EGFR的親和力高于HCP-LCE。第三輪分選后,對12個克隆的VL片段測序檢測EGFR和PD -L1結合。
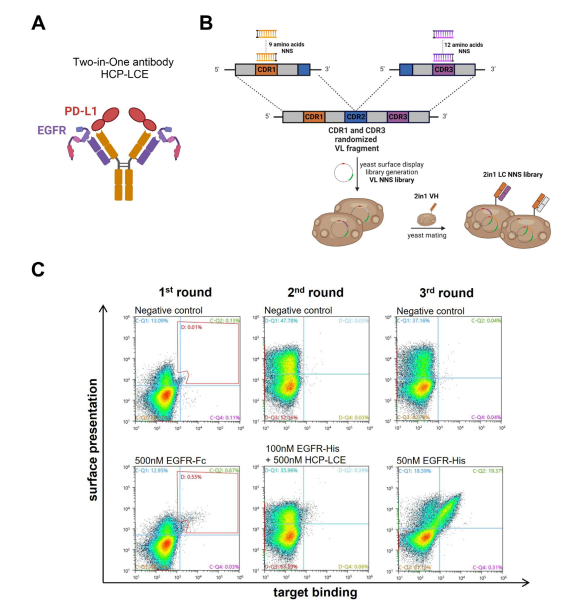
圖1. HCP-LCE輕鏈NNS 酵母表面展示文庫的生成和篩選。(A)EGFR和PD-L1結合的二合一抗體HCP-LCE的示意圖。(B)YSD文庫生成的克隆步驟示意圖。LCDR1和LCDR3的單個氨基酸被編碼20種天然氨基酸的密碼子所取代。通過酵母交配將輕鏈多樣性與野生型HCP-LCE重鏈結合。使用BioRender.com創建。(C)通過流式細胞術對二倍體 HCP-LCE 輕鏈突變 YSD文庫進行篩選。y軸表示利用抗人λ鏈抗體 AF647或PE標記的表面展示,而X軸分別利用抗人Fc PE抗體或抗6×His AF647抗體表示EGFR-Fc或EGFR-His結合。
分析的12個二合一的LC突變體中,多數有野生型 LCDR1 區域,這表明輕鏈CDR1 并非主要參與EGFR結合;所有變異體在 LCDR3 同一位置突變,顯示該位置對EGFR 結合有主要影響,12突變體中,有11個在LDCR3的第3位酪氨酸向谷氨酸突變,也有1有向天冬氨酸突變的。還產生一個攜帶酪氨酸到賴氨酸突變的變體。
Expi293FTM 細胞被共轉染產生全長二合一抗體突變體,突變體經蛋白A親和層析純化。SDS-PAGE分析顯示有預期的重鏈及輕鏈且無降解產物,熱穩定性研究表明HCP-LCE變體熔化溫度在60.5℃至66.5℃之間,野生型抗體的熔化溫度為58.9℃,表明熱穩定性沒有降低(表1)。
表1. HCP-LCE、LCE-E、LCE-D和LCE-K的生物物理性質,包括BLI測量的親和力和動力學結合率、流式細胞術測量的EC50值、PRODIGY web服務器預測的親和力值和熔化溫度。
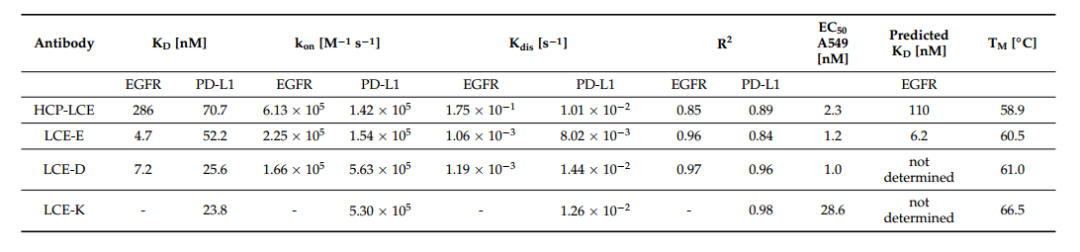
采用BLI測量確定 HCP- LCE 變體對EGFR和PD-L1的親和力(圖2)。突變體 LCE-D和LCE-E對EGFR的親和力比野生型增加了約60倍,KD值在個位數納摩爾范圍(圖2,表1),親和力顯著增加的主要原因是由于解離率提高,而LCE-K突變體不靶向EGFR,表明LCDR3第三位對 EGFR 結合影響較大。所有四種變體對PD-L1結合動力學相似,說明LCDR3突變對PD-L1結合無主要影響,PD-L1的KD值在兩位數納摩爾范圍(表1)。
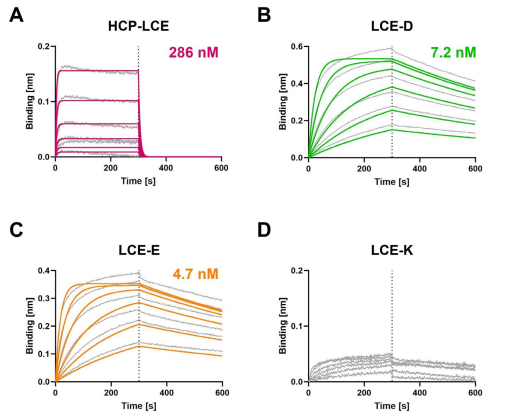
圖2. 通過BLI測量表征HCP-LCE變體的EGFR結合。BLI測定(A) HCP-LCE、(B) LCE-D、(C) LCE-E和(D) LCE-K對EGFR的影響。LCE-D和LCE-E靶向EGFR的親和力高于HCP-LCE,而突變體LCE-K不能結合EGFR。擬合用彩色曲線表示。
HCP-LCE和LCE-E與兩種靶標的結合動力學在helix®生物傳感器儀器上使用switchSENSE®技術進行了表征。DNA共軛靶標與熒光團標記的適配器鏈混合,后者混合在芯片表面上錨定鏈。分析物的結合是由熒光團感知的,熒光團對其局部環境的變化很敏感。在這種情況下,分析物結合導致了猝滅效應,可見為信號降低(圖3A-D)。該技術還可以使用不同的熒光團標記不同的抗原,在雙色實驗中對同一表面上的抗原進行平行分析。此外,比較了兩種抗體變體與EGFR和PD-L1的結合動力學,無論是在一個固定蛋白的表面上,還是在兩個目標蛋白都固定的表面上。觀察到的相互作用表現為兩相解離,且解離速率有快有慢(圖3)。在LCDR3 (LCE-E變體)中引入谷氨酸減少了快速解離速率的影響,并穩定了抗體與EGFR的結合(圖3C)。
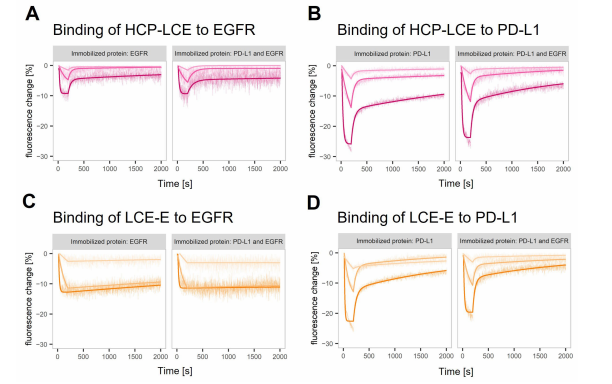
圖3. 實時抗原結合測量單蛋白和雙蛋白表面。(A,C) EGFR和(B,D) PD-L1結合(A,B) HCP-LCE和(C,D) LCE-E在EGFR或PD-L1表面和兩種蛋白表面上的特性(每個圖的左和右面板)。擬合模型假定所有觀察到的相互作用均為雙相解離。在LCE-E與EGFR的相互作用中,快速解離相的貢獻最小。
為驗證HCP-LCE突變體與腫瘤細胞親和力,對EGFR和PD-L1雙陽性A549細胞進行流式細胞儀結合實驗。用不同濃度抗體染色細胞并檢測結合情況。LCE-E和LCE-D顯示特異性細胞結合,EC50值約為1nM,結合最大值相似(圖4)。野生型HCP-LCE親和力低,EC50為2.3nM且最大結合值顯著降低。LCE-K因缺失EGFR結合特性,細胞結合也受影響(圖4A,B)。
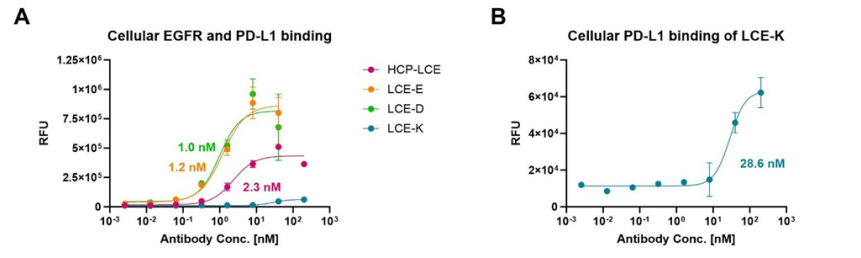
圖4. HCP-LCE變體在EGFR/PD-L1雙陽性A549細胞上的結合。(A) A549細胞上HCP-LCE(粉色)、LCE-E(橙色)、LCE-D(綠色)和LCE-K(藍色)的細胞滴定。(B) A549細胞上LCE-K的細胞滴定;A所示圖的放大視圖。利用變斜率四參數擬合來擬合所得曲線。EC50值:HCP-LCE, 2.3 nM;LCE-E, 1.2 nM;LCE-D, 1.0 nM;LCE-K, 28.6 nM。所有的測量都是重復進行的,實驗至少重復了三次,得到了相似的結果。
利用heliXcyto生物傳感器設計原生分子互作細胞分析系統(RT-IC)實驗,揭示改進的EGFR結合動力學對抗體變體LCE-E與靶陽性腫瘤細胞結合動力學的影響,并與野生型HCP-LCE進行比較。使用兩種不同的EGFR和PDL1雙陽性腫瘤細胞系A431和A549測量結合動力學。在A431細胞中,EGFR和PD-L1的表達水平高于A549細胞,且兩種細胞系中EGFR的表達均高于PD-L1的表達[35]。將細胞裝入heliXcyto生物傳感器芯片上的細胞捕獲網中,在增加抗體濃度的情況下,分三個注射步驟注射熒光標記的抗體變體。抗體的關聯通過熒光信號的增加可見;切換到緩沖流后,信號的減少明顯反映兩者的解離。在A431細胞和A549細胞上檢測到兩種抗體(HCP-LCE和LCE-E)的結合(圖5A-D)。
在大多數情況下,解離速率是雙相的,并且以較慢的解離速率為主(圖6)。以較慢的解離速率(KD2)計算的KD值在A431細胞上的HCP-LCE在0.1-0.7 nM范圍內。HCP-LCE在A431上較低的KD2值是由于更快的結合速率引起的(圖6A,B)。
在兩種細胞系中,LCE-E顯示較慢的第二次解離速率(kd2)(圖6C,D)和較弱的第二次解離相對總解離振幅的貢獻(圖6C)。這導致親和成熟抗體在細胞表面保留的時間增加(圖6D)。
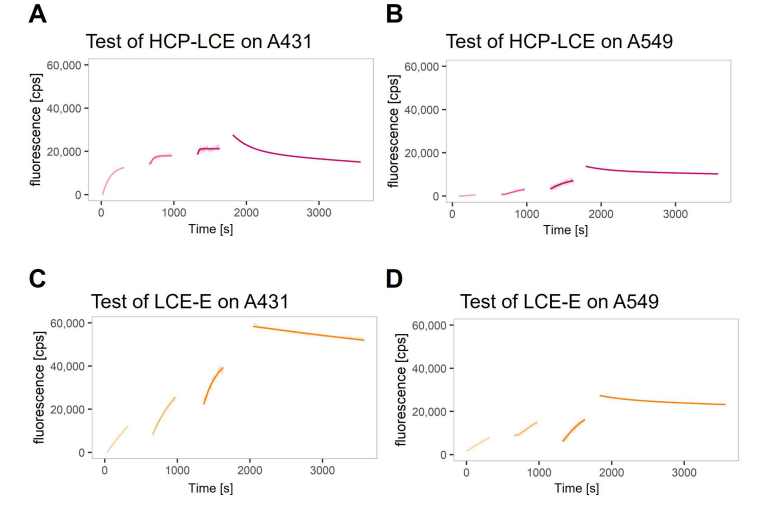
圖5. 在EGFR和PD-L1雙陽性的A431和A549細胞上的實時結合動力學。在helixcyto生物傳感器中通過RT-IC測量的(A、B)HCP-LCE 和(C、D)LCE-E在(A、C)A431和(B、D)A549 細胞上的實時結合曲線。細胞用多聚甲醛固定,并加載到芯片上的五個獨立細胞捕獲網中。不斷增加濃度并分三個連續注射步驟注入熒光標記的分析物。分析物的結合通過熒光信號的上升而明顯顯示出來。在第三次注射后,系統切換到緩沖液流以監測分析物的解離,表現為信號的明顯降低。數據點用假設單相結合和雙相解離的動力學模型進行擬合。
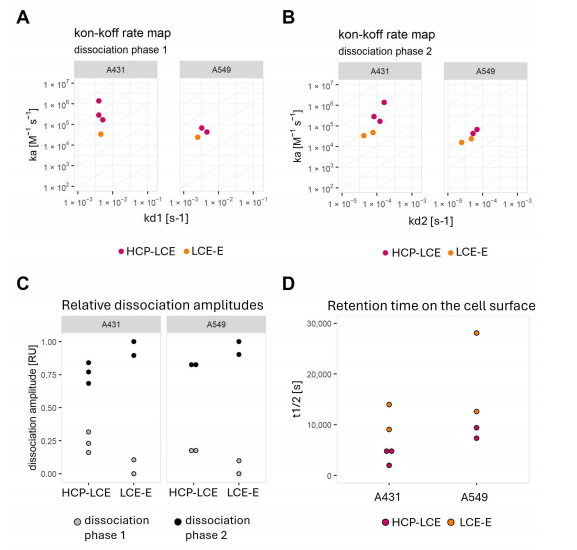
圖6. 用假設分析物的單相結合、雙相解離以及完全解離的動力學模型擬合并結合曲線后計算出的動力學值的圖形。開-關速率圖繪制(A)結合速率與解離速率1的關系,或(B)與解離速率2的關系。(C)展示較快解離速率(解離階段 1)和較慢解離速率(解離階段 2)相對貢獻的圖。(D)根據擬合模型為每種相互作用計算的半衰期值的圖。
為研究LCE-E突變體與野生型HCP-LCE和其抗原的相互作用,使用AlphaFold multitimer[24,25]。結果顯示,PD-L1結合主要通過重鏈CDRs(圖7A),EGFR結合涉及重鏈和輕鏈CDRs(圖7B),且HCDR3和LCDR3主要靶向EGFR表位,與實驗數據一致[23]。PRODIGY web服務器預測蛋白-蛋白復合物的親和力[32],結果顯示 HCP-LCE與EGFR結合KD值為110 nM,LCE-E與EGFR結合KD值為6.2 nM,表明LCDR3氨基酸交換使親和力顯著增加,原因是LCDR3位置3的谷氨酸和EGFR 位置165的精氨酸形成鹽橋所引起(圖7C)。為了證明二合一VH:VL二聚體可以同時靶向EGFR和PD-L1,研究采用了蛋白對接方法[33,36]。PD-L1與EGFR:VH:VL復合物對接,形成一個四聚體蛋白復合物,其中兩種抗原可以同時結合而沒有空間位阻(圖7D)。
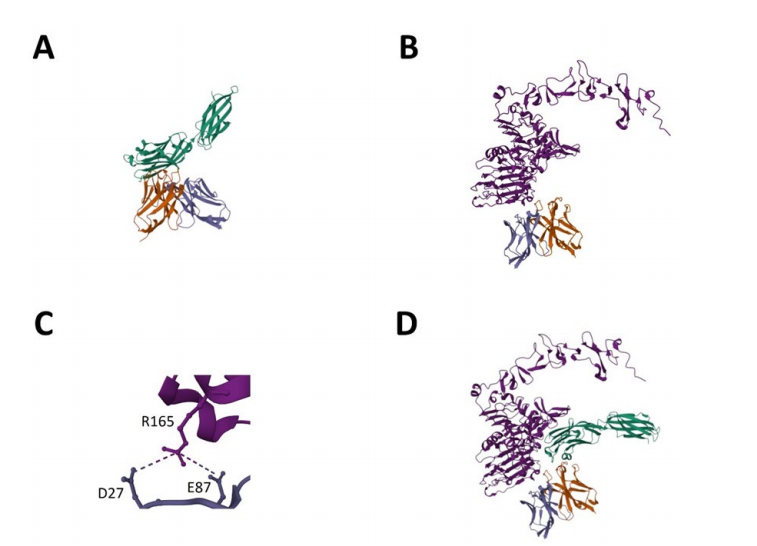
圖7. 基于AlphaFold的LCE-E與EGFR和PD-L1結合的模型。(A)LCE-E與PD-L1 胞外結構域(綠色)的結合。(B)LCE-E與EGFR 胞外結構域(淺紫色)的結合。(C)LCE-E的LCDR3 處的谷氨酸(E87)與 EGFR胞外結構域 165 位的精氨酸(R165)之間的鹽橋。(D)PD-L1與 EGFR:VH:VL 復合物對接的四聚體蛋白復合物。VH 片段顯示為橙色,VL 片段顯示為深紫色。使用AlphaFold Multimer創建。
親和成熟是抗體獲得增強親和性和功能性的過程,由B細胞中免疫球蛋白基因體細胞超突變及抗原結合選擇產生,通常發生在急性感染或接種疫苗后數周內[37]。與種系編碼抗體相比,親和成熟后的抗體高度突變且親和力顯著增加,是蛋白工程中提高體外親和及結合作用、優化抗體治療潛力的關鍵技術[38,39]。該研究生成雞源性EGFR×PD-L1 二合一抗體親和成熟變體以增強 EGFR 結合特性,通過位點飽和誘變,流式細胞儀檢測隨機YSD文庫分離出變體LCE-E,其因LCDR3第三位氨基酸由Y變為E而改善了EGFR親和力,AlphaFold模型表明可能是形成鹽橋所致。
通過多種方法證實二合一變體LCE-E的EGFR結合親和力提高,如BLI測量、混合表面實時抗原結合測量和腫瘤細胞結合實驗等,其解離率降低使結合更穩定。但高親和力不一定保證提高臨床療效,如帕利珠單抗變體雖效力提高了44倍,但體內療效僅出現了適度改善,并且非特異性結合導致藥代動力學譜較差[40]。HCP-LCE是嵌合抗體,由雞源性VH和VL結構域移植到人IgG1支架上[23],雞的重鏈和輕鏈產生與嚙齒類動物不同,禽類V (D) J基因重排時只有小部分免疫球蛋白基因被選擇,通過體細胞基因轉化和超突變進一步多樣化[41,42]。因HCP-LCE由兩只雞免疫產生,重鏈和輕鏈不在同一只雞中,體內未親和成熟,顯示出體外親和優化潛力。HCP-LCE同時靶向EGFR和PD-L1,抑制EGFR信號傳導[23],也有其他研究描述抗原EGFR和PD-L1的有利組合。
總的來說,該研究已經提出了一種直接的方法來實現雞源性二合一抗體的親和力成熟,優化了兩個靶點中只有一個與單個Fab片段同時處理的親和力。對LCDR3區域的單個氨基酸進行突變,然后進行YSD文庫生成和FACS篩選,結果分離出一種靶向EGFR的變異,其親和力增加了60倍。BLI測量表明,親和度的增加主要是由于解離率的提高。switchSENSE®與RT-IC的結果表明,LCE-E中LCDR3引入的谷氨酸降低了對PD- L1結合的快速解離率影響,對EGFR的結合更穩定且 LCE-E在A431和A549 細胞上的解離速率較HCP-LCE更慢,保留在細胞表面的時間更長。
Alphafold的模型預測,LCDR3中的谷氨酸與EGFR形成鹽橋,導致親和力增加。谷氨酸與賴氨酸的交換(LCE-K)完全消除了EGFR的結合特性。目前為止,這代表了第一個親和成熟的二合一抗體,對其兩個靶點之一的結合進行了優化。
該研究提出了一種雞源Two-in-One抗體親和力成熟的簡單方法,優化了單個Fab 片段同時靶向的兩個靶點中的一個靶點的親和力,通過switchSENSE®與RT-IC等多種實驗方法驗證了變體特性。在抗體應用領域中,switchSENSE®可深入探究抗體與抗原的結合動力學,為理解抗體作用機制提供關鍵數據。RT-IC能夠實時監測抗體在靶細胞上的結合情況,有助于評估抗體的靶向性和有效性。二者結合可為抗體的研發、優化和臨床應用提供重要的技術支持和理論依據。

同騰睿杰(上海)生物科技有限公司作為Dynamic Biosensors中國總代理商,為您提供優質的售前售后服務。
聯系電話:021-50826962
聯系郵箱:sales@ttbiotech.com
[1]. Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev.Drug Discov. 2010, 9, 325–338.
[2]. Stanfield, R.L.; Wilson, I.A. Antibody Structure. Microbiol. Spectr. 2014, 2.
[3]. Tsuji, I.; Vang, F.; Dominguez, D.; Karwal, L.; Sanjali, A.; Livengood, J.A.; Davidson, E.; Fouch, M.E.; Doranz, B.J.; Das, S.C.;et al. Somatic Hypermutation and Framework Mutations of Variable Region Contribute to Anti-Zika Virus-Specific Monoclonal Antibody Binding and Function. J. Virol. 2022, 96, e0007122.
[4]. Green, N.S.; Lin, M.M.; Scharff, M.D. Somatic hypermutation of antibody genes: A hot spot warms up. Bioessays 1998, 20, 227–234.
[5]. Tas, J.M.J.; Mesin, L.; Pasqual, G.; Targ, S.; Jacobsen, J.T.; Mano, Y.M.; Chen, C.S.; Weill, J.-C.; Reynaud, C.-A.; Browne, E.P.; et al. Visualizing antibody affinity maturation in germinal centers. Science 2016, 351, 1048–1054.
[6]. Strohl, W.R.; Strohl, L.M. Sources of antibody variable chains. In Therapeutic Antibody Engineering; Elsevier: Amsterdam, The Netherlands, 2012; pp. 77–595, ISBN 9781907568374.
[7]. Bogen, J.P.; Elter, A.; Grzeschik, J.; Hock, B.; Kolmar, H. Humanization of Chicken-Derived Antibodies by Yeast Surface Display. Methods Mol. Biol. 2022, 2491, 335–360.
[8]. Tabasinezhad, M.; Talebkhan, Y.; Wenzel, W.; Rahimi, H.; Omidinia, E.; Mahboudi, F. Trends in therapeutic antibody affinity maturation: From in-vitro towards next-generation sequencing approaches. Immunol. Lett. 2019, 212, 106–113.
[9]. Persson, H.; Kirik, U.; Th?rnqvist, L.; Greiff, L.; Levander, F.; Ohlin, M. In Vitro Evolution of Antibodies Inspired by In Vivo Evolution. Front. Immunol. 2018, 9, 1391.
[10]. Lou, J.; Marks, J.D. Affinity Maturation by Chain Shuffling and Site Directed Mutagenesis. In Antibody Engineering; Kontermann, R., Dübel, S., Eds.; Scholars Portal: Berlin/Heidelberg, Germany, 2010; pp. 377–396, ISBN 978-3-642-01143-6.
[11]. Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317.
[12]. Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557.
[13]. Tsuruta, L.R.; Dos, M.L.; Moro, A.M. Display Technologies for the Selection of Monoclonal Antibodies for Clinical Use. In Antibody Engineering; B?ldicke, T., Ed.; InTech: London, UK, 2018; ISBN 978-953-51-3825-9.
[14]. He, M.; Khan, F. Ribosome display: Next-generation display technologies for production of antibodies in vitro. Expert Rev. Proteomics 2005, 2, 421–430.
[15]. Chowdhury, P.S.; Pastan, I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat. Biotechnol. 1999, 17, 568–572.
[16]. Rajpal, A.; Beyaz, N.; Haber, L.; Cappuccilli, G.; Yee, H.; Bhatt, R.R.; Takeuchi, T.; Lerner, R.A.; Crea, R. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc. Natl. Acad. Sci. USA 2005, 102, 8466–8471.
[17]. Laffly, E.; Pelat, T.; Cédrone, F.; Blésa, S.; Bedouelle, H.; Thullier, P. Improvement of an antibody neutralizing the anthrax toxin by simultaneous mutagenesis of its six hypervariable loops. J. Mol. Biol. 2008, 378, 1094–1103.
[18]. Chen, S.; Li, J.; Li, Q.; Wang, Z. Bispecific antibodies in cancer immunotherapy. Hum. Vaccin. Immunother. 2016, 12, 2491–2500.
[19]. Sheridan, C. Bispecific antibodies poised to deliver wave of cancer therapies. Nat. Biotechnol. 2021, 39, 251–254.
[20]. Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134.
[21]. Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 expression in cancer and clinical implications in immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11.
[22]. Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20.
[23]. Harwardt, J.; Bogen, J.P.; Carrara, S.C.; Ulitzka, M.; Grzeschik, J.; Hock, B.; Kolmar, H. A Generic Strategy to Generate Bifunctional Two-in-One Antibodies by Chicken Immunization. Front. Immunol. 2022, 13, 888838.
[24]. Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; ?ídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
[25]. Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682.
[26]. Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
[27]. Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; ?ídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021.
[28]. Mirdita, M.; Steinegger, M.; S?ding, J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 2019, 35, 2856–2858.
[29]. Steinegger, M.; S?ding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028.
[30]. Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932.
[31]. Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659.
[32]. Vangone, A.; Bonvin, A.M.J.J. PRODIGY: A Contact-based Predictor of Binding Affinity in Protein-protein Complexes. Bio Protoc. 2017, 7, e2124.
[33]. Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852.
[34]. Siloto, R.M.; Weselake, R.J. Site saturation mutagenesis: Methods and applications in protein engineering. Biocatal. Agric. Biotechnol. 2012, 1, 181–189.
[35]. Bogen, J.P.; Carrara, S.C.; Fiebig, D.; Grzeschik, J.; Hock, B.; Kolmar, H. Design of a Trispecific Checkpoint Inhibitor and Natural Killer Cell Engager Based on a 2 + 1 Common Light Chain Antibody Architecture. Front. Immunol. 2021, 12, 669496.
[36]. Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 2022, 31, e4379.
[37]. MacLennan, I.C. Germinal centers. Annu. Rev. Immunol. 1994, 12, 117–139.
[38]. Chan, T.D.; Brink, R. Affinity-based selection and the germinal center response. Immunol. Rev. 2012, 247, 11–23.
[39]. Chan, D.T.Y.; Groves, M.A.T. Affinity maturation: Highlights in the application of in vitro strategies for the directed evolution of antibodies. Emerg. Top. Life Sci. 2021, 5, 601–608.
[40]. Sassi, A.B.; Nagarkar, R.; Hamblin, P. Biobetter Biologics. Novel Approaches and Strategies for Biologics, Vaccines and Cancer Therapies; Elsevier: Amsterdam, The Netherlands, 2015; pp. 199–217, ISBN 9780124166035.
[41]. Kurosawa, K.; Ohta, K. Genetic diversification by somatic gene conversion. Genes 2011, 2, 48–58.
[42]. Mallaby, J.; Mwangi, W.; Ng, J.; Stewart, A.; Dorey-Robinson, D.; Kipling, D.; Hershberg, U.; Fraternali, F.; Nair, V.; Dunn-Walters, D. Diversification of immunoglobulin genes by gene conversion in the domestic chicken (Gallus gallus domesticus). Discov. Immunol. 2023, 2, kyad002.

